Pediatrics
8.1 Leukocoria
Etiology
Retinoblastoma: A malignant tumor of the retina that appears as a white, nodular mass that breaks through the internal limiting membrane into the vitreous (endophytic), as a yellowish subretinal mass lesion often underlying a serous retinal detachment (exophytic), or as a diffusely spreading lesion simulating uveitis (diffuse infiltrating). Iris neovascularization is common. Pseudohypopyon and vitreous seeding may occur. Cataract is uncommon, and the eye is normal in size. May be bilateral, unilateral, or multifocal. Diagnosis is usually made in patients under 5 years of age, with a mean age of 18 months. A family history may be elicited in about 10%.
Toxocariasis: A nematode infection that may appear as a localized, white, elevated granuloma in the retina or as a diffuse endophthalmitis. Associated with localized inflammation of ocular structures, vitreous traction bands and related macular dragging, traction retinal detachment, and cataract. It is rarely bilateral and is usually diagnosed between 6 months and 10 years of age. Paracentesis of the anterior chamber may reveal eosinophils; serum enzyme-linked immunosorbent assay (ELISA) test for Toxocara organisms is positive. The patient may have a history of contact with puppies or of eating dirt. Toxocariasis may also be acquired prenatally and present as a congenital infection.
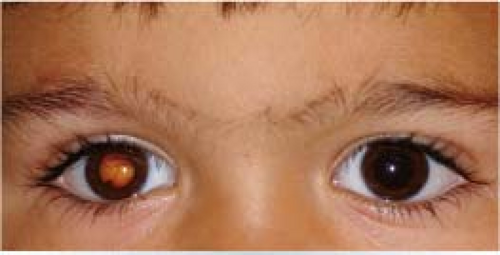
Figure 8.1.1 Leukocoria.
Coats disease (see Figure 8.1.2): A retinal vascular abnormality resulting in small multifocal outpouchings of the retinal vessels. Leukocoria may develop secondary to an exudative, often bullous retinal detachment associated with lipid-rich subretinal fluid or to extensive, yellow intraretinal and subretinal exudate. Usually develops in boys during the first decade of life; more severe cases occur in early childhood. Coats disease is rarely bilateral. No family history.
Persistent fetal vasculature (PFV) (previously known as persistent hyperplastic primary vitreous [PHPV]): A developmental ocular abnormality consisting of a varied degree of glial and vascular proliferation in the vitreous cavity. It is usually associated with a slightly small eye; can also be seen in an eye of normal size or, in cases associated with glaucoma, an enlarged eye.
Typically there is a membrane behind the lens that may place inward traction on the ciliary processes. This is a progressive condition with a cataract present at birth or early in life. The membrane and lens may rotate anteriorly, shallowing the anterior chamber and resulting in secondary glaucoma. Retinal detachments may be seen. Rarely bilateral. No family history.

Figure 8.1.2 Coats disease.
Pediatric cataract: Opacity of the lens present at birth; may be unilateral or bilateral. There may be a family history or an associated systemic disorder. SEE 8.8, PEDIATRIC CATARACT.
Retinal astrocytoma: A sessile to slightly elevated, yellow-white retinal mass that may be calcified and is often associated with tuberous sclerosis and rarely neurofibromatosis. May occur on the optic nerve head (giant drusen) in patients with tuberous sclerosis.
Retinopathy of prematurity (ROP): Predominantly occurs in premature children. Leukocoria is usually the result of a retinal detachment. SEE 8.2, RETINOPATHY OF PREMATURITY.
Others: Retinal detachment, retinochoroidal coloboma, familial exudative vitreoretinopathy (FEVR), myelinated nerve fibers, uveitis, toxoplasmosis, trauma, CMV retinitis, endophthalmitis, retinal dysplasia, incontinentia pigmenti, Norrie disease, medulloepithelioma.
Work-Up
History: Age at onset? Family history of one of the conditions mentioned? Prematurity? Contact with puppies or history of eating dirt?
Complete ocular examination, including a measurement of corneal diameters (look for a small eye), an examination of the iris (look for neovascularization), and an inspection of the lens (look for a cataract). A dilated fundus examination and anterior vitreous examination are essential.
Any or all of the following may be helpful in diagnosis and planning treatment:
B-scan ultrasonography (US), especially if there is no view of the fundus. This can be used to look for calcification within a suspected lesion or a persistent stalk from the nerve to the back of the lens.
Intravenous fluorescein angiogram (useful for evaluation of Coats disease, ROP, retinoblastoma).
Computed tomographic (CT) scan or magnetic resonance imaging (MRI) of the orbit and brain, particularly for bilateral cases of retinoblastoma or those with a family history. Also advised in cases of advanced Coats disease.
Serum ELISA test for Toxocara (positive at 1:8 in the vast majority of infected patients).
Systemic evaluation by pediatrician, especially if concern for retinal astrocytoma or retinoblastoma.
Anterior chamber paracentesis and serum ELISA test for evaluation of toxocariasis (serum antibody test positive at 1:8 in the vast majority of patients infected with Toxocara). SEE APPENDIX 13, ANTERIOR CHAMBER PARACENTESIS.
 NOTE: Anterior chamber paracentesis in a patient with a retinoblastoma can possibly lead to tumor cell dissemination.
NOTE: Anterior chamber paracentesis in a patient with a retinoblastoma can possibly lead to tumor cell dissemination.
May need examination under anesthesia (EUA) in young or uncooperative children, particularly when retinoblastoma, toxocariasis, Coats disease, or ROP is being considered as a diagnosis. If there is increased concern for inherited retinoblastoma, screening examination should be completed within 1 to 2 weeks of life. SEE 8.8, PEDIATRIC CATARACT, for a more specific cataract work-up.
Treatment
Retinoblastoma: Chemoreduction, intra-arterial chemotherapy, intravitreal chemotherapy, cryotherapy, thermotherapy, laser photocoagulation, or plaque radiotherapy. These treatment modalities are typically used in combination. Enucleation is reserved for cases not amenable to the above treatment options. Systemic chemotherapy is used in metastatic disease. Irradiation is rarely used as it is associated with a high incidence of secondary tumors later in life.
Toxocariasis:
Steroids (topical, periocular, or systemic routes may be used, depending on the severity of the inflammation).
Consider a surgical vitrectomy when vitreoretinal traction bands form or when the condition does not improve or worsens with medical therapy.
Consider laser photocoagulation of the nematode if it is visible.
Coats disease: Laser photocoagulation or cryotherapy to leaking vessels. Though associated
with a poor outcome, surgery may be required for a retinal detachment. Consider anti-VEGF agents if significant exudate, subretinal or intraretinal fluid is present.
PFV:
Cataract and retrolental glial membrane extraction.
Treat any amblyopia, though visual outcome is often poor secondary to foveal hypoplasia associated with PFV.
Pediatric cataract: SEE 8.8, Pediatric Cataract.
Retinal astrocytoma: Observation.
ROP: SEE 8.2, RETINOPATHY OF PREMATURITY.
Follow-Up
Variable, depending on the diagnosis. If any concern for heritable disorders, consider referral to a genetic counselor and screening of family members.
8.2 Retinopathy of Prematurity
Risk Factors
Prematurity, especially ≤30 weeks of gestation.
Birth weight ≤1,500 g (3 lb, 5 oz).
Use of supplemental oxygen, neonatal sepsis, hypoxemia, hypercarbia, failure to thrive, and coexisting illness.
Risk factors mentioned above, when present concurrently, have an additive effect on the risk for development of ROP.
Signs
Critical. Avascular peripheral retina. Demarcation line between vascular and avascular retina.
Other. Extraretinal fibrovascular proliferation, vitreous hemorrhage, retinal detachment, or leukocoria, sometimes bilateral. Engorgement and tortuosity of the vessels in the posterior pole and/or iris in plus disease. Poor pupillary dilation despite mydriatic drops. In older children and adults, decreased visual acuity, amblyopia, myopia, strabismus, macular dragging, lattice-like vitreoretinal degeneration, or retinal detachment may occur.
Differential Diagnosis
FEVR: Appears similar to ROP, except FEVR is most commonly autosomal dominant (though family members may be asymptomatic); asymptomatic family members often show peripheral retinal vascular abnormalities. There usually is no history of prematurity or oxygen therapy. SEE 8.3, FAMILIAL EXUDATIVE VITREORETINOPATHY.
Incontinentia pigmenti: X-linked dominant condition that only occurs in girls. Lethal in males. Characterized by skin changes including erythematous maculopapular lesions, vesicles, hypopigmented patches, and alopecia. Associated with eosinophilia. Central nervous system and dental abnormalities may be seen.
SEE 8.1, LEUKOCORIA, for additional differential diagnoses.
Classification
Location
Zone I: Posterior pole: Twice the disc–fovea distance, centered around the disc (poorest prognosis).
 NOTE: With the nasal edge of the optic disc at one edge of the field of view with a 28D lens, the limit of Zone I is at the temporal field of view.
NOTE: With the nasal edge of the optic disc at one edge of the field of view with a 28D lens, the limit of Zone I is at the temporal field of view.
Zone II: From zone I to the nasal ora serrata; temporally equidistant from the disc.
 NOTE: ROP should not be considered zone III until one is sure the nasal side is vascularized to the ora serrata.
NOTE: ROP should not be considered zone III until one is sure the nasal side is vascularized to the ora serrata.
Zone III: The remaining temporal periphery.
Extent

Severity
Stage 1: Flat demarcation line separating the vascular posterior retina from the avascular peripheral retina (see Figure 8.2.1).
Stage 2: Ridged demarcation line.
Stage 3: Ridged demarcation line with fibrovascular proliferation or neovascularization extending from the ridge into the vitreous (see Figure 8.2.2).
Stage 4A: Extrafoveal partial retinal detachment.
Stage 4B: Foveal partial retinal detachment.
Stage 5: Total retinal detachment.
 NOTE: Overall stage is determined by the most severe manifestation; however, it is recommended to define each stage and extent.
NOTE: Overall stage is determined by the most severe manifestation; however, it is recommended to define each stage and extent.
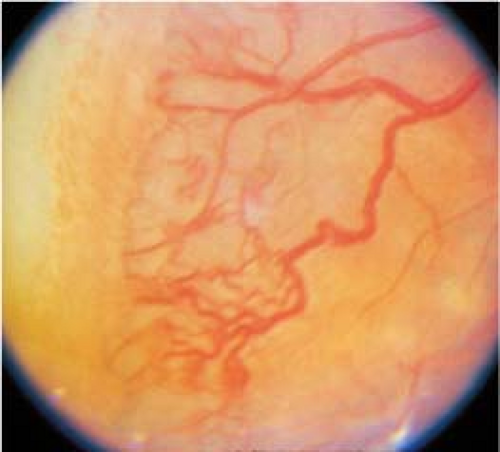
Figure 8.2.2 Retinopathy of prematurity: Stage 3.
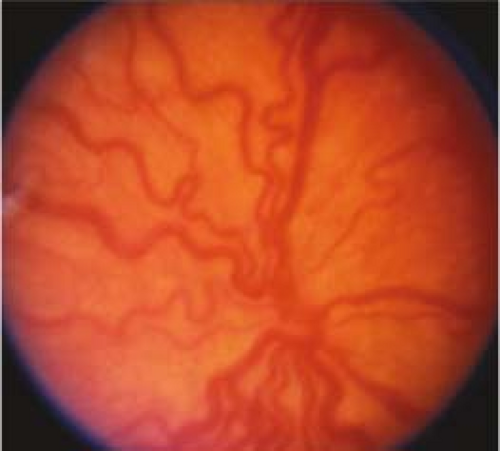
Figure 8.2.3 Retinopathy of prematurity: Plus disease.
“Plus” Disease
At least two quadrants of engorged veins and tortuous arteries in the posterior pole; iris vascular engorgement, poor pupil dilatation, and vitreous haze with more advanced plus disease. If plus disease is present, a “+” is placed after the stage (e.g., stage 3+). If vascular dilatation and tortuosity are present but inadequate to diagnose plus disease, it is called “pre-plus” disease and noted after the stage (e.g., stage 3 with pre-plus disease). Posterior ROP (usually Zone I) with plus disease out of proportion to the peripheral retinopathy, or so-called “rush” disease (also known as aggressive posterior disease), may progress rapidly to stage 5 ROP without passing through the other stages. This aggressive ROP may also show hemorrhages at the junction between vascular and avascular retina (see Figure 8.2.3.).
Type 1 ROP
Defines high-risk eyes that meet the criteria for treatment:
Zone I, any stage with plus disease.
Zone I, stage 3 without plus disease.
Zone II, stage 2 or 3 with plus disease.
Type II ROP
Defines less severely advanced eyes that should be monitored closely for progression to Type 1 disease:
Zone I, stage 1 and 2 without plus disease.
Zone II, stage 3 without plus disease.
Prethreshold and Threshold Disease
Terminology historically used as part of a classification system based on the CRYO-ROP study. Originally determined treatment criteria, but no longer used as part of standard of care.
Screening Recommendations
Birth weight ≤1,500 g.
Gestational age ≤30 weeks.
Selected infants with birth weight >1,500 g or gestational age ≥31 weeks with unstable clinical course thought to be at high risk.
Timing of first eye examination is based on postmenstrual (gestational age at birth plus chronologic age) and postnatal (chronologic since birth) age. The first eye examination should start at 31 weeks postmenstrual age or 4 weeks postnatal age, whichever is later.
 NOTE: The American Academy of Pediatrics (AAP) provides updated guidelines for ROP screening in premature infants. For the latest recommendations, please see their most recent policy statement.
NOTE: The American Academy of Pediatrics (AAP) provides updated guidelines for ROP screening in premature infants. For the latest recommendations, please see their most recent policy statement.
Work-Up
Dilated retinal examination with scleral depression at 31 to 32 weeks after date of mother’s last menstrual period, or 4 weeks after birth, whichever is later.
Can dilate with any two-agent combination from the following: phenylephrine, 1%; tropicamide, 1%; cyclopentolate, 0.2% to 0.5%. Consider repeating the drops in 30 to 45 minutes if the eye is not dilated.
Treatment
Always based on severity and location. Therapeutic goal is ablation of immature, avascular zones of retina. Laser photocoagulation is preferred over cryotherapy. Treatment should be instituted within 48–72 hours (see Figure 8.2.4). Use of intravitreal anti-VEGF agents is an emerging treatment modality, especially when photocoagulation is not available or in very posterior Zone 1 cases; however the long-term effects and potential risks of these medications in preterm infants have yet to be determined.

Figure 8.2.4 Retinopathy of prematurity after laser treatment.
Type 1 ROP needs treatment.
Type 2 ROP should be followed closely.
For stages 4 and 5: Surgical repair of retinal detachment by scleral buckling, vitrectomy surgery, or both is recommended.
Follow-Up
A single ocular examination is sufficient only if it unequivocally shows full retinal vascularization in both eyes.
One week or less: immature vascularization, zone I, no ROP; immature retina localized to boundary of zone I and II; zone I, stage 1 or 2; zone II, stage 3; or any concern for aggressive posterior ROP.
One to 2 weeks: immature vascularization localized to posterior zone II; or zone II, stage 2; or zone I, regressing ROP.
Two weeks: immature vascularization localized to zone II, no ROP; zone II, stage 1; or zone II, regressing ROP.
Two to 3 weeks: zone III, stage 1 or 2; or zone III, regressing ROP.
Children who have had ROP have a higher incidence of myopia, strabismus, amblyopia,
macular dragging, cataracts, glaucoma, and retinal detachment. An untreated fully vascularized fundus needs examination at age 6 months to rule out these complications.
 NOTE: Because of the possibility of late retinal detachments and other ocular complications, ROP patients should be followed at yearly intervals for life.
NOTE: Because of the possibility of late retinal detachments and other ocular complications, ROP patients should be followed at yearly intervals for life.
Acute-phase ROP screening can be discontinued when any of the following signs is present, indicating that the risk of visual loss from ROP is minimal or passed:
Zone III retinal vascularization attained without previous zone I or II ROP. If there is doubt about the zone or if the postmenstrual age is <35 weeks, confirmatory examinations may be warranted.
Postmenstrual age of 50 weeks and no ROP disease equivalent to or worse than zone I, any stage or zone II, stage 3.
Full retinal vascularization in close proximity to the ora serrata (for cases treated with anti-VEGF therapy).
If treated with anti-VEGF, follow-up should be extended due to risk of ROP recurring after 45 weeks.
8.3 Familial Exudative Vitreoretinopathy
Symptoms
Majority are asymptomatic, but patients may report decreased vision.
Signs
(See Figure 8.3.1.)
Critical. Peripheral retinal capillary nonperfusion, most prominently temporally, and may be present for 360 degrees. Bilateral but may be asymmetric. Peripheral retinal vessels have a fimbriated border. Present at birth.
Other. Peripheral neovascularization and/or fibrovascular proliferation at the border of vascular and avascular retina; temporal dragging of macula through contraction of fibrovascular tissue; radial retinal folds; blunting of the ora serrata; vitreous hemorrhage; tractional, exudative, or rhegmatogenous retinal detachment; peripheral intraretinal and subretinal lipid exudation. May present with strabismus or leukocoria in childhood. Cataract, band keratopathy, neovascular glaucoma, or phthisis possible.
 Figure 8.3.1 FEVR with a falciform fold. |
Differential Diagnosis
ROP: Appears similar to FEVR, but there should be no family history, and there should be a history of prematurity. SEE 8.2, RETINOPATHY OF PREMATURITY.
SEE 8.1, LEUKOCORIA, for additional diagnoses in the differential including retinoblastoma, Coats, PFV, incontinentia pigmenti, Norrie disease, X-linked retinoschisis, and peripheral retinal nonperfusion. Positive family history and bilaterality can help distinguish from others.
Etiology
Due to defects in the Wnt signaling pathway. Usually autosomal dominant, though autosomal recessive, and X-linked cases have been reported. No history of prematurity or oxygen therapy. Histopathologically, FEVR is similar to ROP.
Work-Up
History: Positive family history? No history of prematurity or oxygen therapy?
Complete ocular examination, including dilated retinal examination with a slit lamp and a 60- or 90-diopter lens looking for temporal dragging of the macula. Indirect ophthalmoscopy looking for peripheral nonperfusion, fibrovascular membranes, tractional retinal detachment. Fluorescein angiography can be used to evaluate more subtle vascular abnormalities. All family members should also have dilated retinal examinations.
Genetic testing: Common mutations include FZD4, LRP5, TSPAN12, and NDP. LRP5 mutation has been associated with early onset osteoporosis.
Treatment
Laser of peripheral nonperfused retina is sometimes considered if there is documented progression. Small, stable tufts of neovascularization can be observed. Scleral buckling and vitrectomy can be considered for retinal detachments. Often complicated by proliferative vitreoretinopathy. Treat amblyopia as needed. Genetic testing for common mutations may be considered.
Follow-Up
Asymptomatic patients should be followed every 6 to 12 months throughout life to monitor for progression.
8.4 Esodeviations
Signs
(See Figure 8.4.1.)
Critical. Either eye is turned inward. The nonfixating eye turns outward to fixate straight ahead when the previously fixating eye is covered during the cover–uncover test. SEE APPENDIX 3, COVER/UNCOVER AND ALTERNATE COVER TESTS.
Other. Amblyopia, overaction of the inferior oblique muscles, dissociated vertical deviation, and/or latent nystagmus may be present.
Differential Diagnosis
Pseudoesotropia: The eyes appear esotropic; however, there is no ocular misalignment detected during cover–uncover testing. Usually, the child has a wide nasal bridge, prominent epicanthal folds, or a small interpupillary distance (see Figure 8.4.2).
SEE 8.6, STRABISMUS SYNDROMES.

Figure 8.4.1 Esotropia.
Types
Comitant or Concomitant Esotropic Deviations
A manifest convergent misalignment of the eyes in which the measured angle of esodeviation is nearly constant in all fields of gaze at distance fixation.
Congenital (infantile) esotropia: Manifests by age 6 months. The angle of esodeviation is usually large (>40- to 50-prism diopters) and mostly equal at distance and near fixation. Refractive error is usually normal for age (slightly hyperopic). Amblyopia is uncommon but may be present in those who do not cross-fixate. Prohibits development of binocular vision. Family history may be present but is not mandatory. Latent nystagmus, inferior oblique overaction, and dissociated vertical deviation may develop as late findings. Congenital esotropia can occur in up to 30% of
children with neurologic and developmental disorders (e.g., cerebral palsy, hydrocephalus).
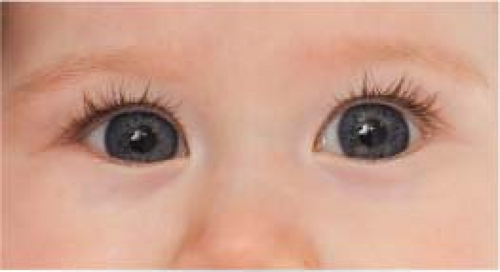
Figure 8.4.2 Pseudoesotropia.
Acquired nonaccommodative esotropia: Convergent misalignment of the eyes not corrected by hyperopic lenses that develops after 6 months of age. Typically starts as intermittent but can become constant over time. Esodeviation is comitant and usually smaller (20 to 35 prism diopters) than that seen in congenital esotropia. Patients may experience diplopia. May be the presenting sign of a brain tumor, however, in most cases there is no associated neurologic or neuroplastic disorder. Usually corrected with strabismus surgery once the angle of the esotropia becomes consistent.
Accommodative esotropia: Convergent misalignment of the eyes associated with activation of the accommodative reflex. May present at 6 months to 6 years of age with the average age of onset being 2.5 years.
Subtypes of accommodative esotropia:
Refractive accommodative esotropia: These children are hyperopic in the range of +3.00 to +10.00 diopters (average, +4.75). The measured angle of esodeviation is usually moderate (20- to 30-prism diopters) and is relatively equal at distance and near fixation. Full hyperopic correction eliminates the esodeviation. The accommodative convergence–accommodation angle ratio (AC/A) is normal. Amblyopia is common at presentation.
Nonrefractive accommodative esotropia (high AC/A ratio): The measured angle of esodeviation is greater at near fixation than at distance fixation. The refractive error may range from normal for age (slight hyperopia) to high hyperopia (may be seen in conjunction with refractive-type accommodative esotropia) or even myopia. Amblyopia is common.
Partial or decompensated accommodative esotropia: Refractive and nonrefractive accommodative esotropias that have a reduction in the esodeviation when given full hyperopic correction, but still have a residual esodeviation. When partial, the residual esodeviation is the nonaccommodative component.
Sensory-deprivation esotropia: An esodeviation that occurs in a patient with a monocular or binocular condition that prevents good vision.
Divergence insufficiency: A convergent ocular misalignment that is greater at distance fixation than at near fixation. This is a diagnosis of exclusion and must be differentiated from divergence paralysis, which, when sudden in onset, can be associated with pontine tumors, neurologic trauma, and elevated intracranial pressure. SEE 10.8, ISOLATED SIXTH NERVE PALSY.
Incomitant or Noncomitant Esodeviations
The measured angle of esodeviation increases in lateral gaze at distance fixation.
Central nervous system pathology causing increased intracranial pressure: Acute and new onset of diplopia secondary to an acquired sixth nerve palsy, which may be accompanied by nystagmus, headache, or other focal neurologic deficits depending on etiology.
Medial rectus restriction (e.g., thyroid disease, medial orbital wall fracture with entrapment).
Lateral rectus weakness (e.g., isolated sixth cranial nerve palsy, slipped or detached lateral rectus from trauma or previous surgery).
Other
Esophoria: Latent esodeviation controlled by fusion. Eyes are aligned under binocular conditions.
Intermittent esotropia: Esodeviation that is intermittently controlled by fusion. Becomes manifest spontaneously, especially with fatigue or illness.
Work-Up
History: Age of onset, frequency of crossing, prior therapy (e.g., glasses, patching).
Visual acuity of each eye, with best correction and pinhole. Color vision and stereopsis.
Ocular motility examination; observe for restricted movements or oblique overactions.
Measure the distance deviation in all fields of gaze and the near deviation in the primary position (straight ahead) using prisms (SEE APPENDIX 3, COVER/UNCOVER AND ALTERNATE COVER TESTS). Look specifically for an esotropia increasing in either side gaze.
Manifest and cycloplegic refractions especially if <7 years of age.
Complete eye examination. Look for any cranial nerve abnormalities and causes of sensory deprivation.
If nonaccommodative esotropia, divergence insufficiency or paralysis, muscle paralysis, or incomitant esotropia develop acutely, an MRI brain and orbit is necessary to rule out an intracranial or orbital process, extraocular muscle pathology, bony lesion, etc.
With incomitant esodeviation greater in side gaze, determine whether the lateral rectus function is deficient or the medial rectus is restricted. Forced duction testing (which may require anesthesia for children) may be necessary for that distinction (SEE APPENDIX 6, FORCED DUCTION TEST AND ACTIVE FORCE GENERATION TEST). Consider thyroid function tests or a work-up for myasthenia gravis. Be sure to look for characteristics of strabismus syndromes (SEE 8.6, STRABISMUS SYNDROMES).
Treatment
In all cases, correct refractive errors of +2.00 diopters or more. In children, treat any underlying amblyopia (SEE 8.7, AMBLYOPIA).
Congenital esotropia: Almost always requires strabismus surgery. However, prescribe glasses and initiate treatment of any underlying amblyopia prior to surgical intervention as appropriate.
Accommodative esotropia: Glasses must be worn full time.
If the patient is <6 years, correct the hyperopia with the full cycloplegic refraction.
If the patient is >6 years, attempts should be made to give as close to the full-plus refraction as possible, knowing that some may not tolerate the full prescription. Attempts to push plus lenses during the manifest (noncycloplegic) refraction until distance vision blurs may be tried to give the most plus lenses without blurring distance vision. The goal of refractive correction should be straight alignment without sacrificing visual acuity.
If the patient’s eyes are straight at distance with full correction, but still esotropic at near fixation (high AC/A ratio), treatment options include the following:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


