Chapter 69 Parathyroid Carcinoma
Introduction
Parathyroid carcinoma is a rare neoplasm that accounts for 0.3% to 5% of all cases of primary hyperparathyroidism (HPT) (see Chapter 70, Surgical Pathology of the Parathyroid Glands). The estimated prevalence of parathyroid carcinoma is around 0.005% of all cancers.1–6 This entity was first described by de Quervain in 1904 in a patient with a nonfunctioning parathyroid carcinoma.7 Subsequently 29 years later in 1933, Sainton and Millot described the first functioning parathyroid carcinoma. Recently, there have been a growing number of clinical reports and case series describing the clinical course, genetics, and the role of different therapeutic modalities in the treatment of this rare disease. Two recent series from Japan and Italy reported the incidence was as high as 5% of all cases of primary hyperparathyroidism. The etiology of this disease is still undetermined, but an underlying genetic foundation is suggested at least in a subset of patients because of the increased incidence in patients with hyperparathyroidism-jaw tumor syndrome.8,9 Head and neck irradiation does not seem to be a major risk factor for parathyroid carcinoma despite individual reports of parathyroid malignancy occurring within an adenoma, hyperplastic gland, or even normal glands after radiation exposure.10–12 Parathyroid carcinoma has also been described in patients with end-stage renal disease and multiple endocrine neoplasia types 1 and 2a,13 thereby raising the possibility that benign hyperplastic parathyroid glands may transform into malignant tumors or that genetic predisposition to disease exists, though this has not been confirmed.10–14
Clinical Presentation
The distinction between benign HPT and malignant disease can be difficult. Frequently, the diagnosis is only made when hypercalcemia reoccurs years later.15 Consideration of carcinoma in the differential diagnosis of parathyroid hormone (PTH)–dependent hypercalcemia has been shown to lead to optimal outcomes, as completeness of resection offers the best chance for cure.4,12,16–21 Clinical and laboratory findings may suggest parathyroid carcinoma; however, these findings are nonspecific (Table 69-1). Several clinical features should, however, alert surgeons to the possibility for this diagnosis and cause them to plan appropriately for a comprehensive resection. In contrast to benign HPT where there is a predominance of females to males (4:1), parathyroid carcinoma affects males and females equally.4,22 In a review of the literature, Koea found that the average age of presentation was 49 years (range 13 to 80 years).22 This is approximately 10 years younger than the average age for benign HPT. Biochemically, the degree of hypercalcemia is more marked in patients with carcinoma than in benign HPT. The average calcium level in benign HPT is 2.7 mmol/L as compared to 3.75 to 3.97 mmol/L reported in the literature for parathyroid carcinoma.86 The PTH levels are also significantly higher, reported to be greater than 5 to 10 times the normal range.3,12,15,23 In a review of reported cases between 1974 and 1998, the mean increase in PTH values was 512% above normal.22 Although such high levels of PTH are seen in progressive secondary and tertiary HPT, primary HPT rarely has such marked elevation.
Table 69-1 The Clinical and Biochemical Features of Parathyroid Carcinoma Compared to Benign Primary Hyperparathyroidism
| Benign HPT | Parathyroid Carcinoma | |
|---|---|---|
| Female to male | 4 to 1 | 1 to 1 |
| Average calcium (mmol/L) | 2.7-2.9 | 3.75-4.0 |
| Average PTH (ng/L) | <2× normal | >3-10 × normal |
| Average age | Sixth decade | Fifth decade |
| Palpable mass | <2% | 30%-76% |
| Osteitis fibrosa cystica | 5% | 40%-75% |
| Nephrolithiasis | 10%-15% | 40% |
| Renal and bone disease | Rare | 40%-50% |
| Asymptomatic | 80% | 2% |
In contrast to benign HPT, where the majority of patients that present today are asymptomatic and lacking in significant manifestations of renal or bone disease,24,25 parathyroid carcinoma patients usually present with clinical manifestations of end-organ disease3,12,22,23 (Table 69-2). The prevalence of renal involvement including, nephrolithiasis and impaired glomerular filtration (GRF) is less than 20% in benign HPT.25,26 In contrast, nephrolithiasis was found in 56% and renal insufficiency in 84% of patients with parathyroid carcinoma.23 Overt radiographic manifestation of HPT, such as osteitis fibrosa cystica, Brown tumors of bone, diffuse osteopenia, and subperiosteal bone resorption, is seen in 45% to 91% of carcinoma patients in contrast to benign HPT where they are seen in only 5% of the population4,22,23,25,26 (Figures 69-2 and 69-3). An additional clinical clue to the diagnosis of parathyroid carcinoma is the presence of both renal and bone disease found in up to 50% of patients. Concomitant new onset renal disease and skeletal manifestations is rarely seen today in benign disease.4,22 Rare cases of nonfunctioning parathyroid carcinoma have been reported.7,27–29 Such tumors tend to present late with diffuse metastatic disease. In a review by Klink, only 14 (2% of all parathyroid carcinomas reported) were nonfunctioning tumors.28
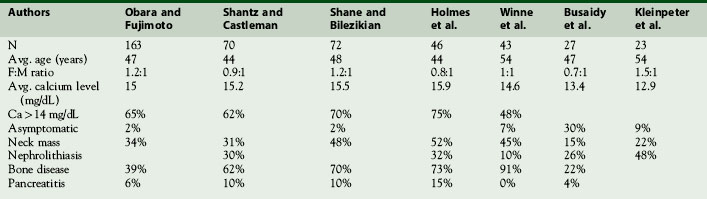
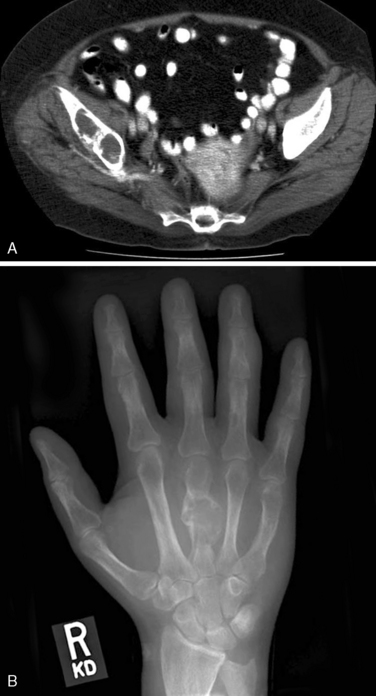
On physical exam, parathyroid cancer patients may display constitutional signs of malignancy such as weight loss, significant fatigue, anorexia, and muscle wasting. The vague nonspecific manifestations of HPT are typically markedly pronounced and unlike benign HPT usually have a rapid onset in patients with carcinoma. A palpable neck mass is found in 36% to 52% of patients with carcinoma and in less than 5% of benign HPT patients.22 Recurrent laryngeal nerve (RLN) palsy in a patient with HPT and no previous neck surgery should be considered highly suspicious for invasive disease. Invasion into the RLN occurs infrequently in 7% to 13% of patients with proved parathyroid cancer. Invasion into surrounding structures was seen in 34% to 75% of patients at the time of operation with the adjacent thyroid gland, overlying strap muscles, and adjacent soft tissues involved most commonly. Tracheal involvement occurs in 11% of patients; the esophagus and carotid sheath each 2% of the time.31
Regional nodal metastases from parathyroid carcinoma are uncommon, with clinical involvement present in only 3% to 8% of cases.30–32 Distant disease was documented in 4.5% of cases in the Surveillance, Epidemiology, and End Results (SEER) database and reported at presentation in smaller series between 4% and 11% of the time.30,31 Twenty-two percent of patients in one series developed distant metastases during long-term follow-up with lung, bone, and liver the most common sites.
Incidence
Parathyroid carcinoma is a rare cause of primary HPT. In 1999 the National Cancer Data Base (NCDB) reported 286 cases of parathyroid carcinoma, the largest single series published to date spanning a 10-year period.6 The researchers found that parathyroid carcinoma accounted for 0.005% of NCDB cancer cases. Beus and Stack reviewed all reports of parathyroid carcinoma in the literature since 1904 and found a total of 711 cases.34 Many of these cases are likely reported multiple times, highlighting the rarity of this unique tumor. Utilizing information from the SEER database, the incidence in the United States is estimated to be 5.73 per 10 million population, increasing by 60% from 1988 to 2003.30 In most series, parathyroid carcinoma accounts for less than 1% of patients presenting with primary HPT.4,15,23,33 Interestingly, there appears to be a higher incidence of this disease in the Japanese and Italian populations, reporting an incidence of 5%.12,35,36 The discrepancy likely arises from either the diagnostic difficulties that face the pathologist in the absence of tumor invasiveness and distant disease, or an absolute increase in the disease resulting from genetic and environmental factors.
Etiology and Molecular Pathogenesis
The etiology of parathyroid carcinoma remains to be fully elucidated. Until recently, clinical associations with predisposing factors such as head and neck radiation, chronic stimulation from renal failure, and familial syndromes were the only clues. Recent advances in molecular genetics, however, have increased our understanding of the pathogenesis of this disease. Several clinical associations that may predispose to the development of parathyroid carcinoma have been reported. Parathyroid carcinoma following head and neck radiation has been reported in a few patients.12,37–41 Three of these patients however, developed parathyroid carcinoma 25, 49, and 53 years following radiation, and a further patient also had chronic renal failure, another possible initiating factor. Therefore, the etiologic significance of head and neck radiation is not clearly understood.
Parathyroid carcinoma has also been described in several patients with chronic renal failure (CRF) on hemodialysis. In a review of the literature, Miki et al. found 12 patients with CRF in whom parathyroid carcinoma developed.42 They outlined the difficulty in distinguishing, clinically, parathyroid carcinoma from progressive secondary HPT in patients with CRF. In none of the reported cases was the diagnosis of carcinoma made preoperatively because of the biochemical and clinical similarities between parathyroid carcinoma and progressive secondary HPT in patients with CRF. With an increase in the number of patients on hemodialysis worldwide, it is interesting that the prevalence of parathyroid carcinoma has remained fairly constant.6 Certainly there can be pathologic difficulty in distinguishing hyperplasia from carcinoma (see Chapter 70, Surgical Pathology of the Parathyroid Glands).
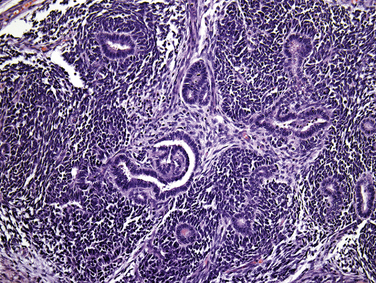
Familial HPT, first reported in 1936, is now known to be a separate entity from multiple endocrine neoplasia type 1 syndrome (MEN 1) (see Chapters 58, Principles in Surgical Management of Primary Hyperparathyroidism, 65, Surgical Management of Multiglandular Parathyroid Disease, 66, Surgical Management of Secondary and Tertiary Hyperparathyroidism, and 67, Parathyroid Management in the MEN Syndromes).43 Familial HPT has an autosomal dominate mode of inheritance. This syndrome is characterized by hypercalcemia, elevated PTH levels, and isolated parathyroid tumors, with no evidence of hyperfunction of any other endocrine tissue. Although multigland hyperplasia is the most common finding, solitary adenomas have been reported in up to 25% of patients.44,45 Parathyroid carcinoma was reported in five of the affected families, leading the authors to conclude that these patients are at increased risk of this rare malignancy.46,48 This clinical syndrome of isolated familial HPT appeared to be distinct from another hereditary HPT syndrome called hyperparathyroidism jaw-tumor syndrome (HPT-JT).46–48 HPT-JT syndrome is a rare autosomal dominant condition associated with ossifying fibromas of the mandible and maxilla, renal cyst, renal hamartomas, and Wilms’ tumors49–53 (Figure 69-1). The incidence of parathyroid carcinoma is found in approximately 10% in HPT-JT syndrome patients compared to 1% in sporadic HPT.46–48,54,55
Since the early 2000s, evidence for the role of both a tumor suppressor gene and an oncogene in the pathogenesis of parathyroid tumors has been reported. The identification of the gene, HRPT2, responsible for HPT-JT syndrome, has been mapped to the 1q25-q32 region.35 Carpten et al. identified 13 different mutations of HRPT2 in 14 families with HPT-JT syndrome. Their study supported the previously held belief that this is a tumor suppressor gene, which encodes for the protein parafibromin. The role of parafibromin is presently unknown. Inactivating germline mutations of this gene were identified in the majority of kindreds with HPT-JT syndrome, in which parathyroid carcinoma is overrepresented; therefore, Shattuck investigated if HRPT2 mutations could be responsible for the development of sporadic parathyroid carcinoma. Ten of the 15 parathyroid cancers studied demonstrated HRPT2 mutations, all of which inactivated the encoded parafibromin protein.56HRPT2 mutation leading to an inactivation of parafibromin is therefore likely an important contributor to the pathogenesis of parathyroid carcinoma. In support of this thesis, Howell also demonstrated HRPT2 somatic mutations in four of four parathyroid carcinomas, and germline mutations in five HPT-JT tumors and two additional tumors from a familial HPT kindred.9 These data provided further evidence for HRPT2 as the causative gene in HPT-JT and a subset of familial HPT. Both these studies support the hypothesis that HRPT2 mutation is an early event that may lead to the development of parathyroid carcinoma (see Chapter 70, Surgical Pathology of the Parathyroid Glands).
Unexpectedly, three patients in Shattuck’s study of sporadic parathyroid carcinoma were found to have germline mutations.56 This finding suggests that some of the sporadic carcinomas may be a phenotypic variant of HPT-JT syndrome. The clinical implications of this finding are significant. Patients, especially the young, should be considered for DNA testing to look for germline mutations of the HRPT2 gene. If mutations are found, then family members can be tested, and, if positive, appropriate surveillance can be implemented.18,56
Further evidence to support the important role that HRPT2 gene plays in the pathogenesis of parathyroid carcinoma is the finding of an increased loss of heterozygosity (LOH) at chromosome 1q in sporadic carcinomas. In Haven’s study that included 22 parathyroid carcinomas, 12 (55%) showed LOH of chromosome 1q, whereas LOH is only found in 8% of parathyroid adenomas.54,57 This is not the entire story, however, as this group also found that 50% of parathyroid carcinomas demonstrated LOH of chromosome 11q13, the location of the MENIN gene. This gene is responsible for multiple endocrine neoplasia type 1 (MEN 1) syndrome, an autosomal dominate disorder characterized by multiple parathyroid adenomas, pituitary, pancreatic neuroendocrine, carcinoid, and adrenal cortical tumors. It has been previously reported that 30% of cases of parathyroid adenomas demonstrate LOH in chromosome 11q13,58,59 suggesting that the MENIN gene may play a role in tumorigenesis. Of interest was that 36% of the parathyroid carcinomas in Haven’s study demonstrated LOH in both chromosomes 1q and 11q13. This finding suggests that inactivation of the HRPT2 gene may function both independently and in concert with the MENIN gene inactivation to promote parathyroid carcinogenesis.54
PRAD1/Cyclin D1 has also been implicated in the development of parathyroid tumors.60–63PRAD1/Cyclin D1 is an oncogene located at chromosome 11q13; its protein product is thought to play a significant role in transition of cells from G1 phase of the cell cycle into the S phase.60 The PRAD1/Cyclin D1 oncoprotein is overexpressed in 18% to 40% of parathyroid adenomas.54,60,62,63 Overexpression of this protein was found in a significantly higher frequency in parathyroid carcinomas (57% to 91%).54,62 Although these data suggest an important role of this protein in parathyroid carcinomas, the genetic effect of PRAD1/Cyclin D1 activation in the context of the development of parathyroid carcinoma remains to be determined.
One possible mechanism for the oncogenic activity of PRAD1/Cyclin D1 is via the inactivation of the growth inhibitory effects of the protein product of the retinoblastoma (RB) gene.8RB is a tumor suppressor gene that has been implicated in parathyroid carcinomas. Several investigators have demonstrated a LOH on chromosome 13q, a region known to include both the RB and the BRCA2 genes.19,64,65 However, Shattuck was unable to demonstrate tumor-specific somatic mutations in either RB or BRCA2 in six parathyroid carcinomas that demonstrated LOH at 13q chromosome.8 Although decreased expression of these genes may contribute to the pathogenesis of parathyroid carcinomas, their role in tumorigenesis needs further investigation.
Pathology
The diagnosis of parathyroid carcinoma remains a challenge for the pathologist (see Chapter 70, Surgical Pathology of the Parathyroid Glands). Frozen section does not allow the distinction of benign from malignant parathyroid tumors, and incisional biopsies are not recommended as they carry the risk of implanting tumor cells into the surrounding tissues.
Until recently the diagnosis was made on the histologic findings of invasion and metastatic spread. These unequivocal features of malignancy, however, are often absent in individual cases of clinically suspected parathyroid carcinoma. Immunohistochemical ancillary tests are now routinely utilized to confirm the diagnoses of parathyroid carcinomas and atypical adenomas in many pathology laboratories.66
Macroscopic and Microscopic Features
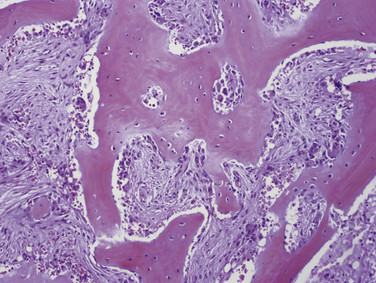
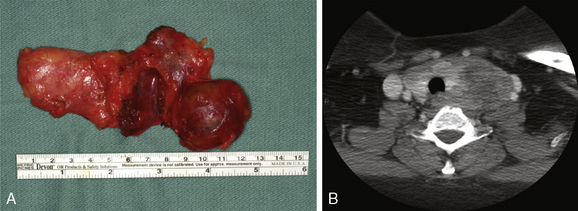
Parathyroid carcinoma can grossly mimic an adenoma. Carcinomas, however, are generally firm and are adherent to the adjacent structures (Figure 69-4). The cut surfaces are gray-white and may show focal necrosis.6,67 The median tumor size is 3.3 cm and the mean weight is 2.7 g (with a range of 0.08 to 13 g) (see Chapter 70, Surgical Pathology of the Parathyroid Glands).6,68
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


