Paragangliomas are highly vascular, slow-growing neoplasms that can occur within the temporal bone, as well as other sites in the head and neck. They develop from paraganglia, which are neurosecretory structures derived from the neural crest. Paraganglia are distributed in the middle ear and jugular bulb but are also found in the carotid and aortic bodies, where they have an established chemosensory function. Paragangliomas are distinguished by their extreme vascularity, which facilitates their diagnosis yet complicates their management. Although typically benign, these tumors can cause significant morbidity by bone erosion, cranial nerve paralysis, and intracranial invasion.
Paraganglia were first described as glomus body-like structures in the carotid body by Von Haller (
1) in 1762, but it was not until 1840 that Valentin (
2) identified these structures in the temporal bone, using the term
ganglionum tympanicum. In 1878 Krause (
3) described a carotid body-like structure in the tympanic canaliculus. Similar structures were also identified in the nodose ganglion and subsequently in a number of sites in the head and neck, including the nasal cavity, the orbit, and the larynx. In 1941 Guild (
4) provided the first modern description of temporal bone paraganglia, and in 1945 Rosenwasser (
5) reported a vascular tumor of the hypotympanum that he concluded must have originated from the
glomus jugularis described by Guild.
Glenner and Grimley (
6) classified paraganglia into four categories: (i) branchiomeric, (ii) juxtavagal, (iii) aorticosympathetic, and (iv) visceral autonomic. Within the branchiomeric category they included jugulotympanic, intercarotid, subclavian, laryngeal, coronary, aorticopulmonary, and pulmonary subtypes. From a clinical standpoint, however, the primary distinction needs to be made among paragangliomas arising from the middle ear (glomus tympanicum), the jugular bulb (glomus jugulare), the vagus nerve (glomus vagale), and the carotid body (carotid body tumor or glomus caroticum). Although in strict terms the designation
glomus tumor should be reserved for neoplasms of glomus bodies contained in skin, the term
glomus in conjunction with an identifying site is well understood in the literature to indicate a paraganglioma.
Several schemes for surgical classification of paragangliomas have been devised. Although one scheme has not been universally applied, the Fisch system is widely accepted, logical, and easily remembered (
Table 41.1) (
7).
EPIDEMIOLOGY
Paragangliomas of the head and neck occur with an approximate incidence of 1 in 30,000. Overall, carotid body tumors are the most common, comprising 60% of all head and neck paragangliomas. However, in the temporal bone the glomus jugulare tumor is the most common, followed by the glomus tympanicum tumor. The relative frequency of tumor sites within the temporal bone reflects the distribution of normal paraganglia as quantified by Guild. In his study of 88 temporal bones, about 50% were found in the adventitia of the jugular bulb, 20% in the inferior tympanic canaliculus, and 10% on the cochlear promontory (
4).
Approximately 10% of all head and neck paragangliomas are familial, but in some populations this proportion has been estimated as high as 50% (
8). Multicentricity occurs in about 10% of all sporadic paragangliomas cases. However, in familial cases, the rate of multicentricity ranges from 25% to 55% (
9).
Paragangliomas are more commonly found in Caucasians and in populations living at high altitude. In addition, there is a striking threefold to sixfold (or greater, depending on the series) predominance of female patients with paragangliomas, despite an equal complement of temporal bone paraganglia between women and men in Guild’s study. Tumors are rarely detected before age 18 years, and most patients are diagnosed in the fifth to sixth decade. Typically, there is a delay in diagnosis of several years from the onset of symptoms. Patients with familial paragangliomas can often be diagnosed in the second or third decade due to heightened awareness and proactive screening.
PATHOGENESIS
Important clues to the pathogenesis of paragangliomas include (i) familial occurrence, (ii) extreme vascularity, (iii) association with hypoxic states, and (iv) an increased incidence in women. Therefore any mechanism postulated for their pathogenesis should account for these clinical features. An important impediment to progress in understanding the pathogenesis of paragangliomas has been the lack of a reliable animal model, despite efforts such as tumor implantation into the subrenal capsule of nude mice (
10).
Familial cases account for at least 10% and possibly a much larger percentage of patients with head and neck paragangliomas (
11). The genes for familial paragangliomas, termed PGL1 and PGL2, are transmitted in an autosomal dominant manner; therefore each child of an affected individual will have a 50% chance of inheriting the gene. However, only individuals who have inherited the gene from their father will eventually develop tumors, demonstrating greater expression with increasing age (
8). This occurs through the process of genomic imprinting, where an allele is altered (possibly by DNA methylation) based on the sex of the transmitting parent (
12). It has been postulated that PGL is inactivated during oogenesis and reactivated in spermatogenesis. Therefore “skipped” generations can occur, because transmitting fathers who have inherited the gene from their mothers will not develop tumors.
Through linkage studies using large Dutch kindreds, PGL1 was originally mapped to two regions of the long arm of chromosome 11, one between loci 22.3 and 23.2 (PGL1) (
13) and another at locus 13.1 (PGL2) (
14). In screening North American kindreds, PGL1 appeared to be the more common (
15,
16). Using these and Dutch kindreds, Baysal and coworkers (
17) made a breakthrough discovery by demonstrating point mutations in
SDHD, a gene that encodes the small subunit protein (cybS) of cytochrome B in mitochondrial complex II. These mutations were shown to result in either premature stop codons that could block synthesis of cybS protein or amino acid substitutions that could alter cybS conformation and function (
17). Interestingly, no evidence for imprinting of
SDHD was found, and the reasons for the discrepancy remain unclear. Because electron transport by mitochondrial complex II has been proposed as a mechanism for oxygen sensing in chemoreceptor organs such as the carotid body, mutations in
SDHD may explain paraganglioma development as a consequence of inappropriate signaling of hypoxia (
17).
Hypoxia has been implicated in the development of these tumors both in animals and humans. For example, bovines experiencing chronic hypoxia due to altitude are predisposed to hyperplasia and subsequently neoplasia of the carotid bodies (
18). Brachycephalic breeds of dogs, including the boxer and the Boston terrier, are predisposed to obstructed airways and also have an increased incidence of paragangliomas of the aortic body (
19).
In humans exposed to chronic hypoxia due to high altitude, the incidence of carotid body tumors increases progressively with higher altitude; ultimately the incidence is about 10 times greater than populations at sea level (
20). There may be differences between paragangliomas that develop at high altitude versus sea level in terms of a greater female predominance and a lower rate of multicentricity and familial tumors (
21). Hyperplasia of the carotid body occurs as a result of chronic hypoxia due to conditions such as emphysema, cyanotic congenital heart disease, and cystic fibrosis (
22,
23). Some investigators have argued that carotid body tumors generated at high altitude are in fact examples of extreme hyperplasia, not neoplasia. However, because hyperplasia can be the precursor of actual neoplasia, these hypoxic states may provide a permissive environment for the development of paragangliomas. Interestingly, patients with hypoxia secondary to chronic obstructive pulmonary disease may be at increased risk for paragangliomas (
24).
Not surprisingly, comparisons have been drawn between PGL and the causative gene for Von Hippel-Lindau disease (VHL), in which a negative regulator of hypoxia-inducible genes is disrupted (
25). In VHL, a number of highly vascular tumors develop, including renal cell carcinomas, central nervous system hemangioblastomas, retinal angiomas, endolymphatic sac tumors, and sometimes paragangliomas
(
26,
27). However, VHL mutations have been examined in a large pedigree of familial paragangliomas, with no mutations found (
28).
The influence of hormones such as estrogens has also been implicated in the development of paragangliomas. Many clinical series show a clear preponderance of female patients by a ratio of 3:1, and the highest disparity reported is 19:1. The disparity is best documented in carotid body tumors: At sea level there is a 2:1 female-to-male ratio, but in high-altitude populations, this ratio increases to 12:1 (
20,
29). In a large clinical series of paragangliomas from multiple sites in the head and neck, the female-to-male ratio was 6:1 (
30). Interestingly, many of the patients in this series may have lived at high altitude. The magnified female-to-male ratio in populations at high altitude suggests an interaction between hypoxia and estrogens in the genesis of these lesions.
Expression of angiogenic peptide growth factors might represent a common mechanism triggered by these external influences. It is established that angiogenesis is a critical step in the progression of solid tumors (
31). Cell-to-cell signaling by angiogenic growth factors, including vascular endothelial growth factor (VEGF), platelet-derived endothelial cell growth factor (PD-ECGF), and basic fibroblast growth factor (bFGF), is essential to the angiogenic process. In a rat model of chronic hypoxia induced by hypobaric conditions, exuberant angiogenesis and hyperplasia of the carotid body was accompanied by a dramatic upregulation of messenger RNA for VEGF and one of its receptors, flk-1 (
32). Furthermore, there is evidence that paragangliomas of the temporal bone and other sites in the head and neck express both VEGF and PD-ECGF protein (
33). Because the expression of VEGF and PD-ECGF are both upregulated by hypoxia and estrogens, synthesis of these growth factors by paragangliomas can contribute to the increased incidence and progression of these tumors in hypoxic states and in women. Expression of these angiogenic growth factors by paragangliomas again draws comparison to Von Hippel-Lindau disease, because many of the hypervascular tumors associated with VHL express hypoxia-inducible factors such as VEGF (
27). Finally, the recently identified mutations in
SDHD may cause inappropriate signaling of hypoxia, which culminates in overexpression of proteins such as VEGF. In addition to driving angiogenesis, which would indirectly support tumor growth, VEGF might interact with its receptors on tumor cells as part of an autocrine (self-sustaining) mechanism (
33).
HISTOLOGY
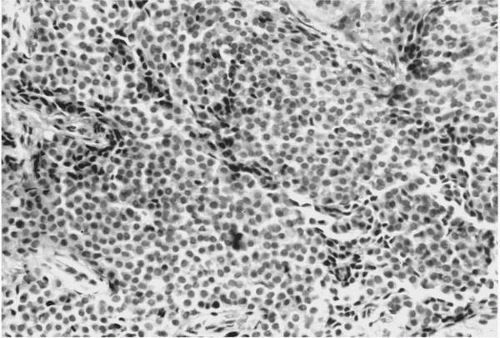
As seen in light microscopy, paragangliomas recapitulate the endocrinologic organization of normal paraganglia. These tumors consist of organoid clusters of type I (“chief”) and type II (“sustentacular”) cells, which have been described as “Zellballen.” These clusters are surrounded by a highly vascular stroma containing numerous capillary-sized vessels (
Fig. 41.1). Chief cells are polygonal, epithelioid cells that comprise the bulk of the Zellballen and are considered to be modified neurons. Their cytoplasm
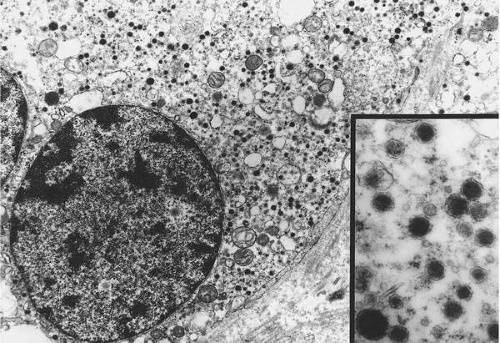
has a granular quality due to the presence of numerous vacuoles and catecholamine-containing granules, which are best demonstrated with electron microscopy (
Fig. 41.2). Elongated sustentacular cells envelop the chief cells but are difficult to observe on standard light microscopy. Their function may be analogous to Schwann cells or pericytes, and a decrease in their number relative to normal paraganglia is a feature of paragangliomas (
34). Identification of sustentacular cells is important, because their presence allows differentiation of paragangliomas from other neuroendocrine lesions such as carcinoid tumors (
35). Most jugulotympanic paragangliomas have a highly cellular pattern; in contrast, hyalinized collagenous septae separating clusters of tumor cells are associated with glomus vagale lesions (
6).
Immunohistochemically, chief cells can be identified by chromogranin staining, whereas sustentacular cells are highlighted by neural markers such as S-100 or neuron-specific enolase. A host of substances, including substance P, vasoactive intestinal peptide, and somatostatin, have also been identified in paragangliomas with immunohistochemistry (
36).
The overall rate of malignancy in paragangliomas is about 5%, considering all sites in the head and neck, and the incidence of malignancy in temporal bone lesions is slightly less (
37). The highest rate of malignancy has been associated with glomus vagale lesions, up to 19% (
38). The exact criteria for “malignancy” have been debated. For example, extensive local invasion and bone destruction have been considered indicators of malignancy. There is also immunohistochemical evidence that malignant paragangliomas express fewer neuropeptides than benign tumors (
36). However, it is now accepted that histologic features such as mitotic figures and nuclear pleomorphism do not reliably distinguish malignant from benign paragangliomas. Even frank DNA aneuploidy is not correlated with either aggressive local extension or metastatic disease (
39). Ultimately, the only clear indicator of malignancy in these lesions is metastatic spread to lymph nodes or distant sites that do not contain paraganglia, such as lung, bone, and liver. Using this criterion, metastases can be easily distinguished from multicentric lesions, which should be presumed if the coexisting lesions are found in sites known to contain paraganglia. It should be noted that evidence of malignancy in the form of bone metastases has been reported up to 6 years after surgical removal of the primary tumor (
40).
Cranial nerve involvement by paragangliomas occurs through infiltration along neural microvasculature, not simple compression. A grading system has been proposed based on involvement of epineurium, perineurium, and endoneurium (
41). Unfortunately, clinical assessment of cranial nerve palsies underestimates the rate of neural invasion observed intraoperatively and confirmed with histopathology; clinically, the facial nerve is most often impaired, whereas intraoperatively the vagus nerve is most often invaded (
41).
CLINICAL PRESENTATION
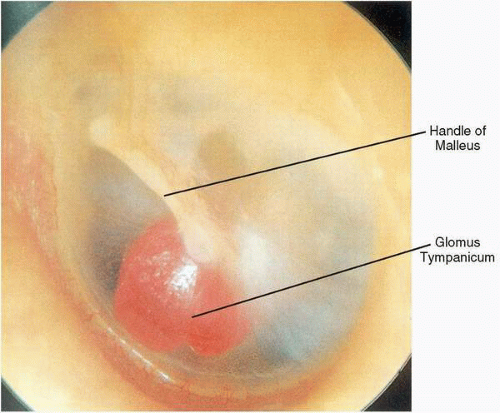
Glomus tympanicum tumors typically present with pulsatile tinnitus and unilateral hearing loss, which is usually conductive in nature. However, sensorineural hearing loss accompanying these lesions has been observed, which occasionally improves after surgical removal. Otoscopic examination reveals a reddish mass, often in the anteroinferior quadrant of an otherwise normal middle ear, forming a meniscus where the mass contacts the tympanic membrane. However, because the tumors are roughly spherical, the meniscus underestimates the size of the tumor. Occasionally, extension of a glomus tympanicum into the eustachian tube orifice results in secondary serous otitis media. When the entire circumference or at least maximum diameter of the mass is visible above the tympanic annulus, the diagnosis of a glomus tympanicum can be made (
Fig. 41.3). With positive pressure pneumatoscopy, Brown’s sign can be elicited, consisting of an initial phase of blanching (by displacement of venous congestion), followed by arterial pulsations (
43).
Glomus jugulare tumors are more varied in presentation due to their capacity to involve lower cranial nerves and to extend intracranially. Nonetheless, otologic symptoms predominate early, with lower cranial nerve deficits occurring later. Pulsatile tinnitus is the most common presenting symptom, followed by conductive hearing loss (
44). Symptoms of lower cranial nerve dysfunction, including hoarseness, dysphagia, aspiration, or dysarthria, are the next most common form of presentation, followed by otalgia and either bloody or purulent otorrhea. Bleeding from the ear can be provoked by coughing, sneezing, or straining. Vertigo can be a presenting symptom in about 20% of cases of glomus jugulare. Presentation with a neck mass is uncommon.
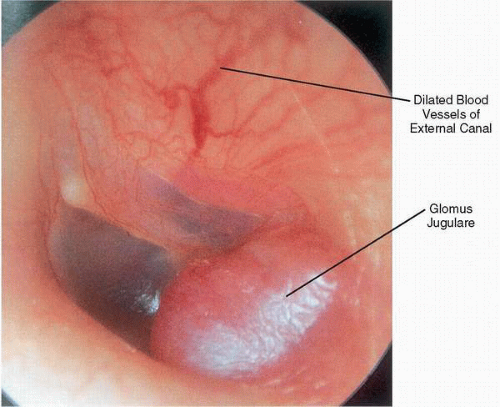
On otoscopy, the appearance of a glomus jugulare tumor can range from the barely visible dome of a vascular mass to complete tumor obliteration of the middle ear space with erosion through the tympanic membrane and formation of a polyp. With any glomus jugulare tumor, the maximum diameter of the mass is hidden below the fibrous annulus. Neovascularization in the form of dilated arterial vessels can be seen along the external auditory canal and tympanic membrane, demonstrating the angiogenic nature of these lesions (
Fig. 41.4).
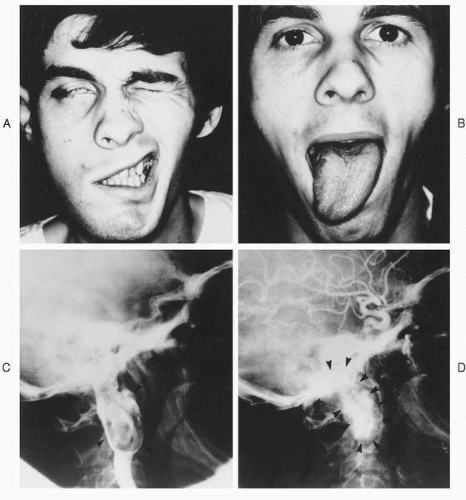
A thorough cranial nerve exam may demonstrate additional palsies that have not troubled the patient, but in most cases the exam will merely confirm symptomatic deficits. On exam, the most common cranial nerve involved by glomus jugulare tumors is the facial nerve (
Fig. 41.5), with the most common site of involvement being the vertical segment (
41). Deficits of cranial nerves IX, X, XI, and XII can occur in isolation or various combinations, and classic jugular foramen syndromes can occur, which are easily recognized. Unfortunately, tumors can also invade these nerves without clinical signs, creating a dilemma when discovered intraoperatively. Deficits of cranial nerves V and VI are unusual, due to the infrequency of middle fossa extension (
45). Involvement of the adventitia of the carotid artery can present as Horner’s syndrome.
By themselves, symptoms and signs cannot reliably predict the extent of glomus jugulare tumors, and it is well known that very extensive tumors can occur with minimal symptoms and sometimes no demonstrable cranial nerve deficits. However, Spector and colleagues (
45) have identified findings that are associated with intracranial extension. In their series, 50% of patients with Horner’s syndrome had evidence of middle fossa invasion, 50% of patients with jugular foramen symptoms had posterior fossa invasion, and 75% of patients with hypoglossal nerve paralysis had posterior fossa extension (
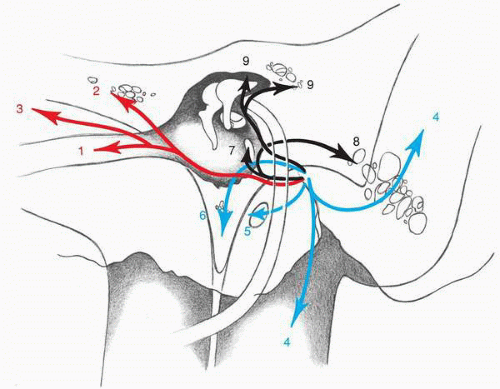
44). Patterns of temporal bone invasion have been described by Spector and colleagues (
45) and are illustrated in
Fig. 41.6.
Facial nerve paralysis can be a presenting symptom of a paraganglioma, most commonly due to involvement of the mastoid segment by direct extension of a glomus jugulare tumor (
41). Rarely, a primary paraganglioma of the fallopian canal, which produces smooth expansion of the canal similar to a schwannoma, can present with a facial palsy or pulsatile tinnitus without facial paralysis (
46). As with most neoplastic involvement of the facial nerve, the paralysis is usually progressive in onset, but it can occur suddenly or even be heralded by twitching or hemifacial spasm.


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
