This article reviews some of the otolaryngologic manifestations of skeletal dysplasias. Achondroplasia is discussed most comprehensively. Skeletal dysplasias are bone and cartilage disorders that disrupt the development of the long bones, craniofacial skeleton, and vertebral column, with the most notable characteristic being short stature. Children with skeletal dysplasias have various medical problems. These children often develop head and neck manifestations of their disorders. Hearing loss, middle ear disease, and respiratory difficulties are seen in these children. Otolaryngologists must be knowledgeable about these disorders to diagnose, treat, and appropriately refer children with skeletal dysplasias.
- •
Children with skeletal dysplasias often present for otolaryngology evaluation; knowledge of the various syndromes and manifestations assists in diagnosis and management of ear, nose, and throat disease.
- •
Children with achondroplasia commonly have middle ear disease from eustachian tube dysfunction, and about half of adults and one-quarter of children with this skeletal dysplasia have hearing loss.
- •
Children with skeletal dysplasias are at high risk for development of obstructive sleep apnea. Although adenotonsillectomy is often the first-line treatment, the causes of sleep-disordered breathing in these patients are complex and the management is difficult.
- •
Children with skeletal dysplasias often undergo a variety of surgical procedures. They present unique anesthetic management issues because of variations in upper and lower airway anatomy, neck motion and stability issues, difficulties with chest and pulmonary mechanics, and abnormalities of neuromotor tone.
| Name of Dysplasia Online Mendelian Inheritance in Man Number ( omim.org ) | Causative Gene Inheritance Pattern | Clinical Characteristics |
|---|---|---|
| Achondroplasia #100800 | FGFR3 AD | Most common skeletal dysplasia. Short limbs mostly in proximal segments, macrocephaly. Midfacial hypoplasia, hypotonia, and obstructive and central apnea. Middle ear dysfunction and hearing loss common |
| Apert syndrome #101200 | FGFR2 AD | Craniosynostosis, midfacial hypoplasia, prognathism, high-arched palate with crowded teeth, syndactyly, choanal atresia, hearing loss |
| Atelosteogenesis types I, II, III Types I and III #108720 Type II #256050 | Type I and III FLNB AD Type II SLC26A2 AR | Atelosteogenesis is a severe disorder of cartilage and bone development. Infants born with this condition have short arms and legs, a narrow chest, and a prominent, rounded abdomen. Cleft palate, clubfeet, and hitchhiker thumbs. They are often stillborn or die soon after birth from respiratory failure. 3 variants recognized currently: types I and III caused by mutation in gene encoding filamin B, type II caused by SCL26A2 and allelic with diastrophic dysplasia |
| Campomelic dysplasia #114290 | SOX9 AD | Severely bowed legs with disorders of the cardiac, respiratory, urinary, and central nervous systems. Manifestations include laryngotracheomalacia, micrognathia, cleft palate, low-set ears, absence of olfactory bulbs, and hypoplastic larynx |
| Cartilage hair hypoplasia #250250 | RMRP AR | A rare form of dwarfism that is characterized by short limbs; fine, sparse hair; impaired immunity; and anemia. More common among certain ethnic groups, particularly the Amish |
| Chondrodysplasia punctata variants #215100, #300180, #302960 | PEX7 AR ARSE X-linked EBP X-linked dominant | Form caused by PEX7 is rhizomelic chondrodysplasia punctata. X-linked form known as CDPX1 or caused by ARSE. CDPX2 also known as Conradi-Hunermann or Happle syndrome caused by X-linked dominant mutations in EBP. Characterized by punctate calcification of the cartilage of the epiphyses, thyroid cartilage, larynx, and trachea. Growth retardation, shortening of limbs, cataracts, dry and scaly skin, nasomaxillary hypoplasia, and mixed hearing loss |
| Cleidocranial dysplasia #119600 | RUNX2 AD | Delayed or absent closure of anterior fontanelle with bulging calvaria, clavicular hypoplasia or aplasia, wide pubic symphysis, unerupted and supernumerary teeth, progressive mixed hearing loss |
| Diastrophic dysplasia #222600 | DTDST/SLC26A2 AR | A form of dwarfism that is characterized by short limbs, cleft palate, clubfeet, hitchhiker thumb, and ears with a cauliflower appearance |
| Ellis-van Creveld syndrome #225500 | EVC AR | Short-stature disorder characterized by short-limb dwarfism, additional fingers and/or toes, abnormal development of fingernails and teeth, and congenital heart defects. Also more common in Amish |
| Hypochondroplasia #146000 | FGFR3 AD | Similar to achondroplasia, but features are less prominent. Often not diagnosed until 2–4 y of age |
| Kniest dysplasia #156550 | COL2A1 AD | Individuals with Kniest dysplasia have round, flat faces with bulging and wide-set eyes. Cleft palate may be present. Tracheomalacia in some infants. Severe myopia is common, and retinal detachment is seen. Hearing loss resulting from recurrent ear infections is also possible. Similar to patients with SED(C) plus additional features of prominent joints and perhaps greater risk of retinal detachment |
| Larsen syndrome #150250 | FLNB AD | Results from resistance to growth hormone. Characterized by prominent forehead, hypertelorism, dislocations of large joints, cleft palate, and abnormalities of the hands and feet. Laryngomalacia/tracheomalacia and subglottic stenosis have been described |
| Multiple epiphyseal dysplasia #132400 | COMP, COL9A1, COL9A2, COL9A3, or MATN3 AD | Typically manifests later in the first decade or older with pain in hips, knees, and ankles. Mildly short stature or average stature |
| Osteogenesis imperfecta #166200, 166210, 166220, #259420 | COL1A1, COL2A2 AD CRTAP, LEPRE1, PPIB, FKBP10, SERPINH1 and SP7 AR | Disorder of abnormal type 1 collagen formation andprocessing, causing bone to be susceptible to fractures. May have average height but a spectrum of severity is seen. Additional signs/symptoms include blue sclera, hypermobile joints with bowing of legs and arms, and hearing loss that can be sensorineural, conductive, or mixed. Phenotypic spectrum varies from few fractures in average stature to lethal |
| Pseudoachondroplasia #177170 | COMP AD | Often average height/length and proportionate at birth, with declining linear growth velocity by 2 y of age. Joint laxity, cervical instability |
| Spondyloepiphyseal dysplasia congenita #183900 | COL2A1 AD | Short stature associated with hearing loss, clubfeet, cleft palate, myopia, odontoid hypoplasia, laryngotracheomalacia, and barrel chest |
| Stickler syndrome #108300, #604841 | Type 1: COL2A1 Type 2: COL11A1 Type 3(nonocular): COL11A2 Type 4: COL9A1 AD | A progressive disorder of collagen (type I, IX, or XI), primarily associated with otologic, ocular, and orthopedic concerns. Eye concerns include myopia, retinal detachment, glaucoma, and strabismus. Other features are recurrent otitis media and sensorineural hearing loss, cleft palate often with the Pierre Robin triad, bifid uvula, flat cheeks and nasal bones, hypermobile joints, scoliosis, and osteoarthritis |
Although skeletal dysplasias primarily manifest with short stature and orthopedic symptoms, not all are associated with short stature and many have associated otolaryngologic and even serious multisystem disease. These patients are best evaluated by a multidisciplinary team of specialists experienced in skeletal dysplasias, and head and neck manifestations of these disorders often require an otolaryngologist.
Head and neck manifestations
The skeletal dysplasia syndromes have a variety of head and neck manifestations. Otolaryngology evaluation of affected individuals can reveal obvious and less obvious ear, nose, and throat disease. The major issues of concern for most children with skeletal dysplasias involve the ears and hearing, and upper airway/respiratory function. An otolaryngologist experienced in the care of syndromic patients can provide specialized diagnostic services and surgical treatment of head and neck disorders associated with skeletal dysplasia. In some cases, otologic or upper airway respiratory symptoms may be the first presenting signs of these conditions.
Achondroplasia
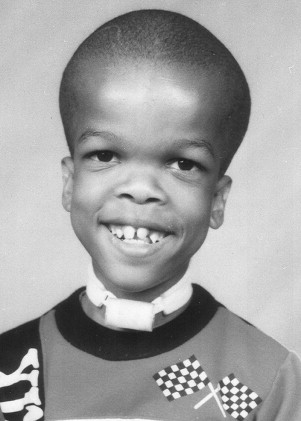
Achondroplasia is the most common syndrome of short-limb dwarfism. This autosomal dominant condition is caused by mutations in fibroblast growth factor receptor 3 (FGFR3). Typical facial features include frontal bone prominence and midfacial hypoplasia, and short stature with rhizomelia is seen ( Fig. 1 ). About 80% of children with achondroplasia are born to parents of average stature, indicating a new mutation in the affected child. Final height for adults with achondroplasia is slightly more than 1.2 m. Although motor delays are common in young children with achondroplasia because of macrocephaly in combination with axial and appendicular hypotonia, cognitive delays are not and require additional evaluation when identified. Almost 40% of children with achondroplasia had conductive hearing loss, and similarly 40% eventually underwent adenotonsillectomy, in one multicenter retrospective study of medical complications of achondroplasia. Otolaryngologic manifestations of achondroplasia are addressed later in this article.
Achondroplasia
Achondroplasia is the most common syndrome of short-limb dwarfism. This autosomal dominant condition is caused by mutations in fibroblast growth factor receptor 3 (FGFR3). Typical facial features include frontal bone prominence and midfacial hypoplasia, and short stature with rhizomelia is seen ( Fig. 1 ). About 80% of children with achondroplasia are born to parents of average stature, indicating a new mutation in the affected child. Final height for adults with achondroplasia is slightly more than 1.2 m. Although motor delays are common in young children with achondroplasia because of macrocephaly in combination with axial and appendicular hypotonia, cognitive delays are not and require additional evaluation when identified. Almost 40% of children with achondroplasia had conductive hearing loss, and similarly 40% eventually underwent adenotonsillectomy, in one multicenter retrospective study of medical complications of achondroplasia. Otolaryngologic manifestations of achondroplasia are addressed later in this article.
Otolaryngologic evaluation of children with skeletal dysplasias
Otolaryngologic symptoms associated with skeletal dysplasia may arise early in the neonatal period or may occur later in childhood. Otolaryngology care involves evaluation of symptomatic children with skeletal dysplasias as well as participation in routine anticipatory longitudinal medical care. The American Academy of Pediatrics Committee on Genetics has recommended, in a guideline published in 1995 and updated in 2005, that children with achondroplasia receive yearly hearing screens from infancy and undergo speech and language evaluation before 2 years of age. This guideline also recommends routine screening for signs of sleep apnea, with a low threshold for testing with polysomnography and/or referral to sleep specialists. This committee suggested that such routine screening for hearing, speech, and sleep apnea is advisable in all other skeletal dysplasia diagnoses until proved to be unnecessary or additional diagnosis-specific recommendations are published. In addition, children with skeletal dysplasias should be followed longitudinally into adulthood for development of otolaryngologic disease such as hearing loss or sleep-disordered breathing.
Routine Evaluations
Routine otolaryngology evaluation for children with skeletal dysplasias should include a careful history directed at issues of:
- •
Hearing acuity
- •
Presence and frequency of otitis media and/or sinusitis
- •
Details of dynamic and static airway function.
The presence and nature of abnormal airway noises (snoring, stertor, stridor) are documented, and careful history about sleep pattern, snoring, and witnessed apneic events is elicited. Details of feeding and swallowing are discussed, and appropriate height and weight measures are documented using appropriate scales for the dysplasia diagnoses when available. Speech and language milestones and clinical hearing assessments are an important part of this evaluation.
Craniofacial features are examined, with emphasis on:
- •
Nasal patency
- •
Size of mandible and maxilla
- •
Shape of palate
- •
Size of tongue
- •
Geometry of oral cavity and oropharynx.
Appearance of mucous membranes and the size of the tonsils and adenoids are documented. Otomicroscopy can be performed in the office to assess structural tympanic membrane abnormalities that may require surgical treatment. Flexible fiber-optic nasopharyngoscopy can assess nasal patency, nasopharyngeal and hypopharyngeal anatomy, supraglottic dynamics, and vocal cord motion.
Airway Evaluation for Suspected Laryngotracheal Abnormalities
When laryngotracheal abnormalities are suspected on initial evaluation, airway radiographs and/or airway endoscopy (laryngoscopy and bronchoscopy) in the operating room may be required. When severe respiratory symptoms are present, operative evaluation may occur at the time the airway is stabilized. Airway management may include endotracheal intubation, or even tracheotomy in the rare child with severe, long-standing, or multilevel airway obstruction not amenable to other treatment.
Cephalometric radiographs can quantify the abnormalities of craniofacial shape that can cause upper airway obstruction in patients with skeletal dysplasia. Computed tomography (CT) is helpful in assessing midfacial bony abnormalities such as choanal atresia, to fully assess bony sinus anatomy and presence of inflammatory sinus disease in skeletal dysplasia patients with symptoms of sinusitis, and to assess middle and inner ear anatomy through temporal bone images ( Fig. 2 ).
Sleep Apnea Evaluation
Skeletal dysplasia patients with sleep-disordered breathing should be evaluated with multichannel nighttime polysomnography to document the presence and severity of sleep apnea, and to distinguish between central and obstructive apnea. A recent guideline on the use of polysomnography in children before tonsillectomy recommended routine use of preoperative polysomnography for children with craniofacial anomalies. This recommendation includes most, if not all, children with skeletal dysplasias who are being considered for surgical treatment of sleep-disordered breathing.
Otologic Evaluation
Patients with skeletal dysplasia and otologic disease need formal hearing assessment using pure-tone and speech audiometry. Otoscopy should evaluate common forms of middle ear disease, such as middle ear effusion, and uncommon entities such as cholesteatoma, ossicular abnormality, or high jugular bulb. When behavioral hearing tests are insufficient, objective tests such as otoacoustic emission screening and auditory brainstem response testing are used to assess hearing in uncooperative or very young patients.
Airway disorders associated with skeletal dysplasia
Children with skeletal dysplasias may present with a range of respiratory signs and symptoms, from acute respiratory distress in the neonatal period to chronic concerns such as pulmonary insufficiency or sleep-associated airway obstruction. Respiratory symptoms in this population arise from multiple factors, including restrictive lung disease from abnormal thoracic anatomy, upper airway obstruction from craniofacial or pharyngeal anatomic issues, and central apneas from brainstem compression or other central nervous system abnormalities.
Upper airway obstruction associated with skeletal dysplasia is usually multifactorial, with dynamic and static obstruction at multiple levels. Abnormal skull base anatomy can decrease the cross-sectional areas of the nasopharyngeal and hypopharyngeal airway. Mandibular and maxillary hypoplasia, abnormal tongue position and size, and large tonsils and adenoids can all contribute to airway narrowing at the pharyngeal level. Brachycephaly with flattening of the nasal dorsum can cause nasal airflow limitation. Associated laryngotracheal anomalies can cause fixed or dynamic narrowing of the large airways in the neck and chest, resulting in recurrent crouplike illness, stridor, and/or exercise intolerance ( Fig. 3 ). Central nervous system disorders may cause poor pharyngeal tone or disorders of ventilatory drive that worsen the respiratory problems caused by the fixed airway obstructions.
Obstructive Sleep Apnea Syndrome in Skeletal Dysplasias
Obstructive sleep apnea syndrome (OSAS) is the most common airway disturbance seen in children with skeletal dysplasias. Children with achondroplasia are at extremely high risk for development of OSAS. OSAS in these children may also have confounding effects on stature through the inhibition of the growth hormone axis. Lanfranco and colleagues observed a reduction in growth hormone secretion, reduced insulinlike growth factor concentration, and peripheral growth hormone insensitivity in patients with OSAS. The adverse growth effects of OSAS may be more significant for children with short-stature skeletal dysplasias.
More than 20 years ago, Reid and colleagues studied 26 children with achondroplasia and found loud snoring by history in 58% and OSAS on polysomnography in 35%. Waters and colleagues studied 20 patients with achondroplasia, 15 children and 5 young adults, using overnight polysomnography. All patients had evidence of upper airway obstruction on sleep study, with 75% of the studies diagnostic for OSAS.
Zucconi and colleagues also diagnosed obstructive apnea and/or hypoventilation in 75% of children with achondroplasia studied with polysomnography. In addition, a recent review of 46 children with achondroplasia showed that 34.3% had sleep studies diagnostic of OSAS, of which 27.3% underwent surgical treatment, with 12.3% of those treated requiring persistent treatment with continuous positive airway pressure (CPAP).
Midfacial hypoplasia, nasal and nasopharyngeal narrowing, and abnormal neural control of pharyngeal muscle tone contribute to OSAS in achondroplasia. in 2005, Onodera and colleagues compared children with OSAS with and without achondroplasia using cephalometric analysis, and the factors contributing to OSAS in children with achondroplasia were pharyngeal airway narrowing, retrognathia, increased mandibular plane angle, and increased height of the lower face.
OSAS has also been described in other skeletal dysplasias including metatropic dwarfism, Melnick-Needles syndrome, and mucopolysaccharidoses. Most children with mucopolysaccharidoses (MPS) suffer from OSAS because of excessive storage of unmetabolized mucopolysaccharides. In a recent study of 24 patients using polysomnography, 22 children with the diagnoses of MPS types I, II, III, IV, and VI all had OSAS, and 88% had moderate or severe OSAS. OSAS in children with MPS is attributed to the infiltration of upper airway tissues with glycosaminoglycans, in addition to tonsillar and adenoidal hypertrophy, neuromotor tone abnormalities, and craniofacial structural issues. Children with MPS have a high likelihood of persistent OSAS after adenotonsillectomy. In addition to palatine tonsil hypertrophy, children with MPS may have lingual tonsil hypertrophy contributing to their symptoms.
Adenotonsillectomy is usually the first choice for treatment of OSAS in children, and this surgery is also used for children with skeletal dysplasias such as achondroplasia. Sisk and colleagues reported that only 18% of their series of achondroplastic children with OSAS required additional treatment after adenotonsillectomy, whereas 90% of the children who had adenoidectomy alone required additional surgery. Adenotonsillectomy was reported to help relieve pulmonary hypertension in a 5-year-old child with achondroplasia and severe OSAS.
P earls and P itfalls : Patients with craniofacial abnormalities are at high risk for respiratory compromise after adenotonsillectomy, and postoperative cardiorespiratory monitoring in an inpatient setting is mandatory for such patients.
Postoperative polysomnography is recommended after adenotonsillectomy for OSAS in children with skeletal dysplasias, because OSAS often persists. Patients with persistent OSAS after adenotonsillectomy can be considered for additional surgery based on specific anatomic issues:
- •
Turbinate reduction
- •
Uvulopalatopharyngoplasty
- •
Tongue resection
- •
Tracheotomy.
Craniofacial reconstructive techniques involving mandibular and midfacial advancement and maxillary expansion may improve OSAS in patients with the craniofacial abnormalities seen in skeletal dysplasias. Successful decannulation of patients with tracheostomy was achieved in 2 young children with achondroplasia using midface distraction osteogenesis procedures. CPAP or bilevel positive airway pressure (BiPAP) are nonsurgical treatments of OSAS in complex patients, when OSAS persists after adenotonsillectomy or other surgery or when surgery is contraindicated. Waters and colleagues studied achondroplastic patients with OSAS, 13 of whom were treated with CPAP, and found improvement in polysomnographic measures and, for some cases, improvement in measured somatosensory evoked potentials. Although CPAP or BiPAP are effective treatments of OSAS in complex sleep-related airway obstruction, difficulties with compliance are magnified in young children.
Central apnea is also seen in children with achondroplasia. Children with achondroplasia may have foramen magnum stenosis and/or hydrocephalus. Brainstem compression at the craniocervical junction has been implicated in abnormal airway control seen in these patients. It is thought that this may be a contributing factor to sudden infant death, which occurs in 2% to 5% of these children. Physicians are encouraged to actively screen patients for this anomaly. Infants with achondroplasia should undergo assessment for craniocervical junction abnormalities, an assessment that often includes careful neurologic history and examination, neuroimaging, and polysomnography. When such compression is identified, surgical decompression of the posterior fossa has improved respiratory function. Although in one small retrospective series up to 50% of patients required decompression of foramen magnum stenosis, a recent review suggests that 5% to 10% of achondroplastic patients require this neurosurgical intervention.
Laryngotracheal Abnormalities in Skeletal Dysplasias
Abnormalities of the larynx and trachea have been associated with specific skeletal dysplasia diagnoses. Myer and Cotton described 2 patients with spondyloepiphyseal dysplasia (SED), an autosomal dominant skeletal dysplasia arising from a mutation affecting collagen II production, who each required tracheotomy for subglottic stenosis. One of the patients with SED had glottic and supraglottic abnormalities in addition to the stenosis below the vocal cords. One patient underwent laryngotracheal reconstruction using a costal cartilage graft, but remained tracheotomy dependent despite surgical efforts.
We have treated 2 such patients with SED and subglottic stenosis requiring tracheotomy in infancy. One underwent costal cartilage graft laryngotracheal reconstruction, but was not decannulated until 4 years after surgery. Another child with SED and congenital subglottic stenosis was decannulated from tracheotomy at age 7 years without surgical intervention.
Myer and Cotton caution that intrinsic cartilage abnormalities in such skeletal dysplasias may compromise the results of standard techniques of augmentation/repair of laryngeal stenosis, which rely on the use of rigid cartilage grafts to restore laryngeal lumen size and framework rigidity.
Long-segment tracheobronchial stenosis has been described in patients with chondrodysplasia punctata, a group of heterogeneous skeletal disorders characterized by punctiform calcifications of bone, in which lengthy areas of tracheal narrowing are associated cartilage calcifications ( Fig. 4 ). Larsen syndrome is a rare skeletal dysplasia characterized by:
- •
Prominent forehead.
- •
Hypertelorism.
- •
Dislocations of large joints.
- •
Abnormalities of the hands and feet.
- •
Laryngomalacia and tracheomalacia have been described in infants.
- •
Congenital subglottic stenosis has been described in 3 patients, all of whom required tracheotomy early in life. These patients were successfully treated by cricotracheal resection with primary anastomosis after failure of augmentation laryngotracheal reconstruction.



