This review describes important aspects of the most commonly encountered craniofacial syndromes. The goal is to provide otolaryngologists and other health care providers with critical information necessary to manage these patients appropriately. The algorithm provided in this article should be helpful in guiding the treatment of craniofacial patients based on their unique otolaryngologic characteristics. The principles highlighted in the algorithm can be applied to other craniofacial syndromes not addressed here, including Pierre Robin sequence and Down syndrome.
- •
A multidisciplinary approach is paramount in the management of the craniofacial patient.
- •
The first step in management of this patient population is ensuring a safe airway.
- •
Nasal obstruction and obstructive sleep apnea are serious potential problems in this patient population.
- •
Evaluation of most craniofacial patients should include a flexible nasopharyngoscopy that can identify choanal atresia, submucous clefts, velopharyngeal insufficiency, nasopharyngeal depth, vocal cord function, and laryngeal clefts and webs.
- •
A conservative approach is needed in patients with cleft palate or submucous cleft when performing an adenoidectomy, as it can lead to velopharyngeal insufficiency.
- •
Craniofacial patients typically require multiple corrective surgeries, performed in staged fashion, requiring computed tomography scan and/or magnetic resonance imaging in preoperative planning.
- •
It is essential to consider and address:
- ○
Hearing loss
- ○
Otitis media
- ○
Social and psychological implications of craniofacial abnormalities
- ○
Challenges with endotracheal intubation and mask ventilation
- ○
Presence of aspiration and gastroesophageal reflux.
- ○
The algorithm provided in this article should be helpful in guiding the treatment of craniofacial patients based on their unique otolaryngologic characteristics. Many aspects of care for this patient population are the same, regardless of the disease process. The principles highlighted in the algorithm can be applied to other craniofacial syndromes not addressed here, including Pierre Robin sequence and Down syndrome, discussed in articles elsewhere in this issue.
General concepts in management
Airway
In otolaryngology, the first step in managing a patient with a craniofacial syndrome is ensuring that he or she has a safe airway; this may start with positioning or other supportive measures. However, some patients require a surgical airway. In one craniofacial clinic, nearly 20% of the 251 craniofacial patients enrolled required a tracheotomy. Most patients will have a flexible and/or direct laryngoscopy. Addressing the airway often involves ruling out aspiration and treating it appropriately if present. Workup often includes a video swallow and esophagram. Evaluation of speech pathology is important, first for feeding and later for speech and language development.
Pearls & Pitfalls : The first step in managing a patient with craniofacial syndromes is to ensure a safe airway. In addition, the otolaryngologist must be aware of the great potential for nasal obstruction and sleep apnea in craniofacial patients .
Gastrointestinal Problems
Gastrointestinal congenital anomalies or common problems such as reflux should be treated. The otolaryngologist must be aware of the great potential for nasal obstruction and sleep apnea in craniofacial patients. In addition, craniofacial patients have a higher risk of developing common otolaryngologic diseases, including upper airway problems such as rhinitis, sinusitis, or laryngitis.
Hearing Loss
Another common problem in this patient population is hearing loss. In addition to the newborn hearing screen performed in the United States, craniofacial patients need to continue to be followed with serial audiograms because hearing loss is common and may be progressive. Those with chronic otitis media may require tympanostomy tubes. Other causes of hearing loss may necessitate surgical intervention as well. However, assistive hearing devices are recommended for many patients, starting early in life.
Multidisciplinary Approach
A multidisciplinary team approach is paramount in the management of the craniofacial patient population. Patients should undergo a genetic workup to determine the etiology of the malformation and identify other potential associated medical problems. Furthermore, genetic counseling can help provide parents with reasonable expectations related to the care of the patient and help them understand potential implications for other family members. In addition to otolaryngologists, pulmonologists and gastroenterologists also commonly provide care for craniofacial patients. Dentists and orthodontists play a crucial role. Most patients need additional support services, such as case management (social work), psychology or psychiatry, speech pathology, physical therapy, occupational therapy, and other educational services. Because of congenital anomalies associated with specific syndromes, various other pediatric specialists may need to be consulted, including cardiologists, ophthalmologists, neurosurgeons, endocrinologists, urologists, nephrologists, and orthopedic surgeons.
Surgical Anesthesia Challenges
Patients with craniofacial syndromes may require multiple surgeries. Surgeons and anesthesiologists should be aware of the potential challenges these patients may have with general anesthesia. In particular, endotracheal intubation or even mask ventilation may be difficult. Patients with micrognathia or macroglossia may be difficult to intubate. Patients with midface hypoplasia are not generally difficult to intubate, but may be difficult to ventilate. An oral airway can be helpful in these patients. Identifying other vertebral or cardiac problems prior to anesthesia can be paramount. For example, patients with Klippel-Feil syndrome may have fusion of cervical vertebrae with limited neck flexion and/or extension. Patients with Down syndrome are at increased risk of atlantoaxial subluxation. In some of these cases, it may be crucial to limit extension of the neck during surgery, or it may be helpful to obtain plain films or computed tomography (CT) of the cervical spine before performing surgical procedures, to rule out cervical spine instability.
Craniofacial synostosis
Craniofacial synostosis refers to premature fusion of one or more of the cranial sutures and abnormal facial development. The most common syndromic craniofacial synostoses are Crouzon, Apert, and Pfeiffer syndromes. Patients usually have maxillary hypoplasia, poor growth of the midface, and a brachycephaly, or fusion of the coronal suture, which results in the appearance of a wide head ( Fig. 1 ). In general, patients with Apert syndrome will have malformations of the hands and feet, such as syndactyly (baseball-glove appearance). Patients with Pfeiffer syndrome may have milder malformations of the digits, such as wide thumbs or toes. Among craniofacial patients, those with craniofacial synostosis may be the most likely to require a tracheostomy secondary to airway problems, approximately 48%.
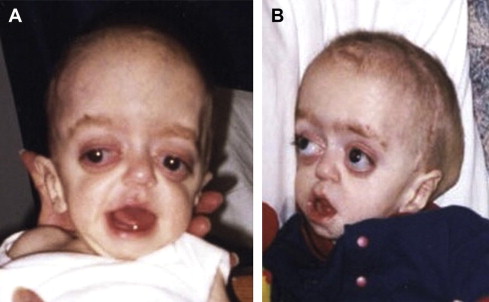
Apert Syndrome: Acrocephalosyndactyly
Background
Apert syndrome was first reported by Wheaton in 1894. However, it was Eugene Apert who expanded its description in 1906. This syndrome refers to the constellation of craniosynostosis, midfacial malformations, and symmetric syndactyly of the hands. Although one of the most severe craniosynostoses, it is fortunately not a common entity. Apert syndrome has been reported in only 15 per 1 million live births.
Apert syndrome is transmitted via autosomal dominant inheritance, although sporadic cases have also been reported. Increasing paternal age is associated with a higher incidence of Apert syndrome. An increased frequency of sperm mutations seems to be implicated. Mutations in fibroblast growth factor receptor 2 (FGFR2) on chromosome 10 are responsible for the autosomal dominant cases of this condition.
Presentation
Apert syndrome can be diagnosed shortly after birth. Common facial features include orbital hypertelorism, proptosis, and down-slanting palpebral fissures. The midface is typically hypoplastic, which causes the mandible to appear protruded. A beaked nose, depressed nasal bridge, and deviated nasal septum are also observed. The nasopharynx is small and the posterior choanae are narrowed. All these features can cause significant nasal obstruction, leading to respiratory difficulty early in life. In addition, the small upper airway predisposes these patients to obstructive sleep apnea (OSA).
Oropharyngeal abnormalities are also commonly noted. A trapezoid-shaped mouth is characteristic. The palate is highly arched with lateral swellings and a median furrow. Cleft soft palate or bifid uvula is found in approximately 75% cases. Eustachian tube dysfunction is also common. Together, these two factors lead to frequent episodes of otitis media.
Hearing loss is frequently seen in patients with Apert syndrome. It is usually bilateral, and conductive hearing loss accounts for up to 80% of cases. Middle ear effusion and a congenitally fixed stapes footplate are common causes of conductive hearing loss in this population. However, middle ear pathology is not the only contributor to hearing loss in these patients. Inner ear malformations, such as a dilated vestibule, malformed lateral semicircular canal, and cochlear dysplasia have also been reported.
Workup
Although the diagnosis of Apert syndrome can be made clinically, a genetic test to detect mutations in FGFR2 is available. Skull and limb radiographs are helpful in detecting specific skeletal abnormalities. CT scanning with 3-dimensional reconstruction can be of paramount value in preoperative planning. In addition, a hearing evaluation is necessary in all patients with Apert syndrome. A flexible nasopharyngoscopy can be useful in assessing the anatomy of choanae and upper airway. Although the diagnosis of OSA can often be made by history and examination alone, polysomnography remains the gold standard.
Management
Positional changes and a nasopharyngeal or oropharyngeal airway may suffice in treating mild obstruction. Nasal secretions should be controlled with gentle suctioning and judicious use of nasal decongestants. Upper respiratory infections should be carefully managed, as they can exacerbate respiratory difficulties. It is also very important to effectively treat otitis media. Although acute infections can be effectively treated with oral antibiotics, chronic middle ear effusions are frequently treated with myringotomy and insertion of ventilation tubes.
In patients with OSA, continuous positive airway pressure may be used to keep the airway open. Tonsillectomy and adenoidectomy may be necessary to help relieve OSA.
Hazard ! If an adenoidectomy is necessary in patients with cleft palate, a conservative surgical approach should be used, given the risk of velopharyngeal insufficiency .
In severely affected children a tracheostomy may be necessary.
Craniofacial reconstruction is performed in a staged fashion. At around 9 to11 months, a cranio-orbital decompression is performed to increase the space in the anterior cranial vault and the orbit. The procedure entails osteotomies of the anterior cranial vault and upper orbits, and bicoronal suture release. This procedure may need to be repeated if the child develops intracranial hypertension. A midline (facial bipartition) split combined with further reshaping of the cranial vault is performed later at age 5 to 7 years. This procedure normalizes the arc of the midface, widens the maxillary arch, and shifts the orbits closer to the midline. The newly assembled midface is advanced horizontally to increase the orbital and zygomatic depth. Le Fort I osteotomies with horizontal advancement must be performed to advance the maxilla and improve malocclusion. A genioplasty may also be performed to help address the lower face deformity.
Crouzon Syndrome: Craniofacial Dysostosis
Background
Crouzon syndrome is characterized by craniosynostosis, proptosis, shallow orbits, and maxillary hypoplasia. This entity occurs in 15 to 16 per 1 million newborns. The main form of inheritance is autosomal dominant with variable penetrance, although sporadic mutations also give rise to some cases. Increasing paternal age seems to play a role. Similarly to Apert syndrome, mutations in FGFR2 are implicated in this syndrome, although the codons involved are different. Recently, a mutation in the transmembrane region of FGFR3 has been observed in these patients.
Presentation
Patients with Crouzon syndrome are characterized by microcephaly, shallow orbits, and a long forehead. The diagnosis may be clear at birth, or may manifest itself later during the first year of life. Ocular proptosis, a feature seen in all the cases, occurs secondarily to the shallow orbits. Cleft lip and palate do not occur frequently. Maxillary hypoplasia leads to a constricted dental arch, which causes the palate to appear high-arched. A beaked nose and a deviated septum can occur, leading to nasal obstruction. Atresia of the nasopharynx and hypopharynx can lead to respiratory distress. The size of the maxillary sinus is significantly reduced. Atresia of the external auditory canals occurs in 13% of cases. Malalignment of the pinna has also been reported. Otitis media and abnormalities of the ossicular chain can occur, and lead to conductive hearing loss. These patients can also develop sensorineural and mixed hearing loss.
Workup
Genetic testing for FGFR2 mutations is available. Most laboratories perform sequence analysis of select exons on the gene, but targeted mutation analysis can also be ordered. Prenatal testing is an option that may be valuable in identifying patients who may have difficulty breathing at birth.
Despite advances in imaging modalities, plain films of the skull remain useful in confirming the diagnosis. Three-dimensional CT scanning remains the best method for delineating the anatomic abnormalities and guiding surgical management. As in patients with Apert syndrome, flexible nasopharyngoscopy can help assess the upper airway. Obstructive sleep apnea can ensue secondary to the midface abnormalities. A polysomnogram is diagnostic. Given the high incidence of hearing loss in these patients, audiologic examination should be performed routinely.
Management
Because of the abnormal midface, nasopharynx, and hypopharynx, one of the main concerns becomes securing the airway from an early age. Some patients will require a tracheostomy shortly after birth. Because many patients undergo staged reconstruction, the tracheostomy may be left in place until all midface surgical procedures are completed.
Similarly to Apert syndrome, multiple reconstructive procedures are necessary, with the first cranio-orbital decompression performed early in infancy at 9 to 11 months unless intracranial hypertension ensues. Later in childhood, at approximately 7 to 9 years of age, the final cranial vault reconstruction is performed and the midface deformity is addressed with a facial bipartition or a Le Fort III osteotomy. A separate Le Fort I osteotomy is needed to restore normal occlusion. This procedure is typically done during adolescence, at 13 to 15 years in females and 15 to 17 years in males. Ancillary procedures such as a genioplasty are performed to address the mandibular deformity.
Finally, because of the different otologic pathologies associated with this syndrome, close otologic and audiologic follow-up is recommended.
CHARGE Association
Background
In 1979, Hall and Hittner reported the first cases of patients with CHARGE association in separate publications. Thus this association is also known as Hall-Hittner syndrome. In 1981, the acronym CHARGE was introduced by Pagon and colleagues. The letters stand for:
Coloboma
Heart defects
Atresia choanae
Retarded growth and development
Genital hypoplasia
Ear anomalies/deafness.
This association of anomalies has an incidence ranging from 0.1 to 1.2 per 10,000. CHARGE is transmitted via an autosomal dominant pattern of inheritance. Mutations in the CHD7 gene, which codes for a DNA helicase, are present in more than 75% of cases. The mutations are usually sporadic, although gonadal mosaicism and familial inheritance have been reported.
Presentation
Patients with the classic 4Cs, namely (1) choanal atresia, (2) coloboma, (3) characteristic ears, and (4) cranial nerve anomalies, are very likely to have CHARGE syndrome. Choanal atresia presents early and dramatically in life, with respiratory difficulty. Atresia may be membranous or bony, and bilateral or unilateral. In some cases, the presence of a cleft lip and palate can substitute for choanal atresia. Patients with CHARGE have flat facies with sloping of the forehead and flattened tip of the nose. The ear is cup-shaped with a hypoplastic lobule, resulting from cartilage deficiency in the outer ear and lack of innervation from the facial nerve to the intrinsic ear muscles. Asymmetric cranial nerve (CN) anomalies can be seen in these patients. Anosmia due to absence or dysfunction of the olfactory nerve (CN I), facial nerve palsy (CN VII), and sensorineural hearing loss (CN VIII) are observed. Furthermore, anomalies attributable to CN IX, X, and XI can lead to swallowing difficulties, esophageal reflux, and velopharyngeal insufficiency. Hearing loss occurs frequently, and can be conductive or sensorineural. In addition to an abnormal CN VIII, patients may have a hypoplastic incus, Mondini defect (abnormal development of the cochlea resulting in less than 2.5 turns), or absent semicircular canal.
Workup
Genetic testing is performed to confirm the diagnosis. Because of the classic middle and inner ear deformities associated with CHARGE, a CT of temporal bone is valuable. An audiogram and auditory brainstem response (ABR) are performed to assess the type and severity of hearing loss. Imaging is very useful in delineating the shape of the choanae. Radiographic visualization of the olfactory tract and other CNs should also be performed.
Management
Bilateral choanal atresia can lead to an airway emergency. As always, stabilizing the airway should be a priority. Endotracheal intubation should be performed in the presence of a pediatric anesthesiologist and/or otolaryngologist. A tracheostomy may be needed in some cases. Surgical correction of choanal atresia can be challenging but should be attempted ( Fig. 2 ). An endoscopic approach can be used first, and a transpalatal approach used if the endoscopic surgery fails. When appropriate, hearing amplification should be started. Cochlear implants may be an option; however, the ear anatomy should be studied and presence of the eighth nerve should be confirmed by CT and/or magnetic resonance imaging (MRI). Speech perception and language outcomes are difficult to assess in patients with developmental delay or other limitations.
Pearls & Pitfalls : Bilateral choanal atresia will often present as an airway emergency .
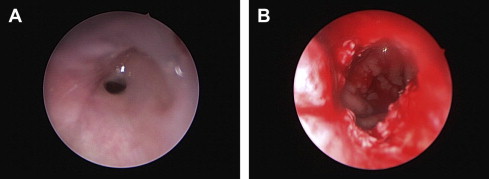
Craniofacial synostosis
Craniofacial synostosis refers to premature fusion of one or more of the cranial sutures and abnormal facial development. The most common syndromic craniofacial synostoses are Crouzon, Apert, and Pfeiffer syndromes. Patients usually have maxillary hypoplasia, poor growth of the midface, and a brachycephaly, or fusion of the coronal suture, which results in the appearance of a wide head ( Fig. 1 ). In general, patients with Apert syndrome will have malformations of the hands and feet, such as syndactyly (baseball-glove appearance). Patients with Pfeiffer syndrome may have milder malformations of the digits, such as wide thumbs or toes. Among craniofacial patients, those with craniofacial synostosis may be the most likely to require a tracheostomy secondary to airway problems, approximately 48%.
Apert Syndrome: Acrocephalosyndactyly
Background
Apert syndrome was first reported by Wheaton in 1894. However, it was Eugene Apert who expanded its description in 1906. This syndrome refers to the constellation of craniosynostosis, midfacial malformations, and symmetric syndactyly of the hands. Although one of the most severe craniosynostoses, it is fortunately not a common entity. Apert syndrome has been reported in only 15 per 1 million live births.
Apert syndrome is transmitted via autosomal dominant inheritance, although sporadic cases have also been reported. Increasing paternal age is associated with a higher incidence of Apert syndrome. An increased frequency of sperm mutations seems to be implicated. Mutations in fibroblast growth factor receptor 2 (FGFR2) on chromosome 10 are responsible for the autosomal dominant cases of this condition.
Presentation
Apert syndrome can be diagnosed shortly after birth. Common facial features include orbital hypertelorism, proptosis, and down-slanting palpebral fissures. The midface is typically hypoplastic, which causes the mandible to appear protruded. A beaked nose, depressed nasal bridge, and deviated nasal septum are also observed. The nasopharynx is small and the posterior choanae are narrowed. All these features can cause significant nasal obstruction, leading to respiratory difficulty early in life. In addition, the small upper airway predisposes these patients to obstructive sleep apnea (OSA).
Oropharyngeal abnormalities are also commonly noted. A trapezoid-shaped mouth is characteristic. The palate is highly arched with lateral swellings and a median furrow. Cleft soft palate or bifid uvula is found in approximately 75% cases. Eustachian tube dysfunction is also common. Together, these two factors lead to frequent episodes of otitis media.
Hearing loss is frequently seen in patients with Apert syndrome. It is usually bilateral, and conductive hearing loss accounts for up to 80% of cases. Middle ear effusion and a congenitally fixed stapes footplate are common causes of conductive hearing loss in this population. However, middle ear pathology is not the only contributor to hearing loss in these patients. Inner ear malformations, such as a dilated vestibule, malformed lateral semicircular canal, and cochlear dysplasia have also been reported.
Workup
Although the diagnosis of Apert syndrome can be made clinically, a genetic test to detect mutations in FGFR2 is available. Skull and limb radiographs are helpful in detecting specific skeletal abnormalities. CT scanning with 3-dimensional reconstruction can be of paramount value in preoperative planning. In addition, a hearing evaluation is necessary in all patients with Apert syndrome. A flexible nasopharyngoscopy can be useful in assessing the anatomy of choanae and upper airway. Although the diagnosis of OSA can often be made by history and examination alone, polysomnography remains the gold standard.
Management
Positional changes and a nasopharyngeal or oropharyngeal airway may suffice in treating mild obstruction. Nasal secretions should be controlled with gentle suctioning and judicious use of nasal decongestants. Upper respiratory infections should be carefully managed, as they can exacerbate respiratory difficulties. It is also very important to effectively treat otitis media. Although acute infections can be effectively treated with oral antibiotics, chronic middle ear effusions are frequently treated with myringotomy and insertion of ventilation tubes.
In patients with OSA, continuous positive airway pressure may be used to keep the airway open. Tonsillectomy and adenoidectomy may be necessary to help relieve OSA.
Hazard ! If an adenoidectomy is necessary in patients with cleft palate, a conservative surgical approach should be used, given the risk of velopharyngeal insufficiency .
In severely affected children a tracheostomy may be necessary.
Craniofacial reconstruction is performed in a staged fashion. At around 9 to11 months, a cranio-orbital decompression is performed to increase the space in the anterior cranial vault and the orbit. The procedure entails osteotomies of the anterior cranial vault and upper orbits, and bicoronal suture release. This procedure may need to be repeated if the child develops intracranial hypertension. A midline (facial bipartition) split combined with further reshaping of the cranial vault is performed later at age 5 to 7 years. This procedure normalizes the arc of the midface, widens the maxillary arch, and shifts the orbits closer to the midline. The newly assembled midface is advanced horizontally to increase the orbital and zygomatic depth. Le Fort I osteotomies with horizontal advancement must be performed to advance the maxilla and improve malocclusion. A genioplasty may also be performed to help address the lower face deformity.
Crouzon Syndrome: Craniofacial Dysostosis
Background
Crouzon syndrome is characterized by craniosynostosis, proptosis, shallow orbits, and maxillary hypoplasia. This entity occurs in 15 to 16 per 1 million newborns. The main form of inheritance is autosomal dominant with variable penetrance, although sporadic mutations also give rise to some cases. Increasing paternal age seems to play a role. Similarly to Apert syndrome, mutations in FGFR2 are implicated in this syndrome, although the codons involved are different. Recently, a mutation in the transmembrane region of FGFR3 has been observed in these patients.
Presentation
Patients with Crouzon syndrome are characterized by microcephaly, shallow orbits, and a long forehead. The diagnosis may be clear at birth, or may manifest itself later during the first year of life. Ocular proptosis, a feature seen in all the cases, occurs secondarily to the shallow orbits. Cleft lip and palate do not occur frequently. Maxillary hypoplasia leads to a constricted dental arch, which causes the palate to appear high-arched. A beaked nose and a deviated septum can occur, leading to nasal obstruction. Atresia of the nasopharynx and hypopharynx can lead to respiratory distress. The size of the maxillary sinus is significantly reduced. Atresia of the external auditory canals occurs in 13% of cases. Malalignment of the pinna has also been reported. Otitis media and abnormalities of the ossicular chain can occur, and lead to conductive hearing loss. These patients can also develop sensorineural and mixed hearing loss.
Workup
Genetic testing for FGFR2 mutations is available. Most laboratories perform sequence analysis of select exons on the gene, but targeted mutation analysis can also be ordered. Prenatal testing is an option that may be valuable in identifying patients who may have difficulty breathing at birth.
Despite advances in imaging modalities, plain films of the skull remain useful in confirming the diagnosis. Three-dimensional CT scanning remains the best method for delineating the anatomic abnormalities and guiding surgical management. As in patients with Apert syndrome, flexible nasopharyngoscopy can help assess the upper airway. Obstructive sleep apnea can ensue secondary to the midface abnormalities. A polysomnogram is diagnostic. Given the high incidence of hearing loss in these patients, audiologic examination should be performed routinely.
Management
Because of the abnormal midface, nasopharynx, and hypopharynx, one of the main concerns becomes securing the airway from an early age. Some patients will require a tracheostomy shortly after birth. Because many patients undergo staged reconstruction, the tracheostomy may be left in place until all midface surgical procedures are completed.
Similarly to Apert syndrome, multiple reconstructive procedures are necessary, with the first cranio-orbital decompression performed early in infancy at 9 to 11 months unless intracranial hypertension ensues. Later in childhood, at approximately 7 to 9 years of age, the final cranial vault reconstruction is performed and the midface deformity is addressed with a facial bipartition or a Le Fort III osteotomy. A separate Le Fort I osteotomy is needed to restore normal occlusion. This procedure is typically done during adolescence, at 13 to 15 years in females and 15 to 17 years in males. Ancillary procedures such as a genioplasty are performed to address the mandibular deformity.
Finally, because of the different otologic pathologies associated with this syndrome, close otologic and audiologic follow-up is recommended.
CHARGE Association
Background
In 1979, Hall and Hittner reported the first cases of patients with CHARGE association in separate publications. Thus this association is also known as Hall-Hittner syndrome. In 1981, the acronym CHARGE was introduced by Pagon and colleagues. The letters stand for:
Coloboma
Heart defects
Atresia choanae
Retarded growth and development
Genital hypoplasia
Ear anomalies/deafness.
This association of anomalies has an incidence ranging from 0.1 to 1.2 per 10,000. CHARGE is transmitted via an autosomal dominant pattern of inheritance. Mutations in the CHD7 gene, which codes for a DNA helicase, are present in more than 75% of cases. The mutations are usually sporadic, although gonadal mosaicism and familial inheritance have been reported.
Presentation
Patients with the classic 4Cs, namely (1) choanal atresia, (2) coloboma, (3) characteristic ears, and (4) cranial nerve anomalies, are very likely to have CHARGE syndrome. Choanal atresia presents early and dramatically in life, with respiratory difficulty. Atresia may be membranous or bony, and bilateral or unilateral. In some cases, the presence of a cleft lip and palate can substitute for choanal atresia. Patients with CHARGE have flat facies with sloping of the forehead and flattened tip of the nose. The ear is cup-shaped with a hypoplastic lobule, resulting from cartilage deficiency in the outer ear and lack of innervation from the facial nerve to the intrinsic ear muscles. Asymmetric cranial nerve (CN) anomalies can be seen in these patients. Anosmia due to absence or dysfunction of the olfactory nerve (CN I), facial nerve palsy (CN VII), and sensorineural hearing loss (CN VIII) are observed. Furthermore, anomalies attributable to CN IX, X, and XI can lead to swallowing difficulties, esophageal reflux, and velopharyngeal insufficiency. Hearing loss occurs frequently, and can be conductive or sensorineural. In addition to an abnormal CN VIII, patients may have a hypoplastic incus, Mondini defect (abnormal development of the cochlea resulting in less than 2.5 turns), or absent semicircular canal.
Workup
Genetic testing is performed to confirm the diagnosis. Because of the classic middle and inner ear deformities associated with CHARGE, a CT of temporal bone is valuable. An audiogram and auditory brainstem response (ABR) are performed to assess the type and severity of hearing loss. Imaging is very useful in delineating the shape of the choanae. Radiographic visualization of the olfactory tract and other CNs should also be performed.
Management
Bilateral choanal atresia can lead to an airway emergency. As always, stabilizing the airway should be a priority. Endotracheal intubation should be performed in the presence of a pediatric anesthesiologist and/or otolaryngologist. A tracheostomy may be needed in some cases. Surgical correction of choanal atresia can be challenging but should be attempted ( Fig. 2 ). An endoscopic approach can be used first, and a transpalatal approach used if the endoscopic surgery fails. When appropriate, hearing amplification should be started. Cochlear implants may be an option; however, the ear anatomy should be studied and presence of the eighth nerve should be confirmed by CT and/or magnetic resonance imaging (MRI). Speech perception and language outcomes are difficult to assess in patients with developmental delay or other limitations.
Pearls & Pitfalls : Bilateral choanal atresia will often present as an airway emergency .



