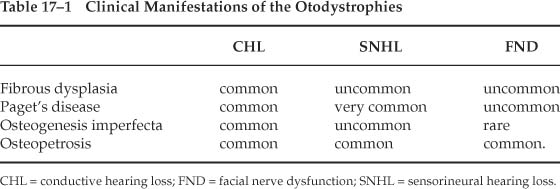
Chapter 17 The otodystrophies are a group of disorders that affect the temporal bone and the otic capsule. Most of these disorders, unlike otosclerosis, affect not only the otic capsule but other areas of the temporal bone as well, in addition to involving other bones of the body. It is important to know that they can present as conductive deafness and that the audiometric configuration can resemble that of otosclerosis. Osteogenesis imperfecta is a rare bone disorder. It represents a group of heterogeneous systemic connective tissue disorders that predispose the individual to recurrent fractures when subjected to trivial trauma. The incidence of osteogenesis imperfecta varies between 2 and 15 per 100,000 births (Pedersen 1985). It is a hereditary disorder of collagen synthesis and occurs in two main forms, osteogenesis imperfecta congenital (OIC) and osteogenesis imperfecta tarda (OIT). In the congenital form, fractures occur in utero, and the fetus dies. In the tarda form, fractures often occur following minor trivial trauma during childhood. This becomes less frequent, however, with the onset of adolescence. Malalignment of the fractured bones leads to deformities and excessive callus formation. OIT has a dominant mode of inheritance, with variable penetrance with asymptomatic carriers, although cases without a family history of OIT are known to occur. The cardinal features of this disease are fractures that occur following relatively minor trivial, often unrecognized trauma. Approximately 50 to 60% of patients eventually develop a hearing loss, and 85% have blue sclera (Quisiling et al 1979). Blue sclerae are also associated with collagen disorders such as Ehlers-Danlos syndrome and Marfan’s syndrome. The symptom complex that includes multiple fractures, blue sclerae, and deafness is known as van der Hoeve’s and de Kleyn’s syndrome. The syndrome was described earlier by Adair Dighton (1912) and Bronson (1917). It was also noticed, however, that in some family members, hearing impairment and blue sclerae presented without the tendency for fractures. Altered collagen synthesis results in defective connective tissue, with a tendency to hypermobility and laxity of joints, thin skin, and easy bruising in the subcutaneous tissues. The teeth of these patients are abnormal in approximately 15% of cases. The dentine is abnormal, or the enamel is cracked, resulting in yellow-stained, irregular teeth. This is known as amelogenesis imperfecta. The clinical course of OIT is variable. Sillence et al (1979) introduced a classification based on genetic and clinical criteria. Type 1 is autosomal dominant, mild, and manifested by blue sclerae. Type 2 is autosomal dominant or recessive and is uniformly lethal. Type 3 is autosomal recessive and is marked by progressive skeletal deformities, with extreme fragility of bones and very short stature. Type 4 is autosomal dominant and is intermediate with patients exhibiting skeletal fragility and short stature. Hearing loss has long been recognized as a common feature in osteogenesis imperfecta and other otodystrophies (Table 17–1). The data in some publications could be taken to imply that with advancing age, the number of patients suffering from osteogenesis imperfecta approaches 100%. The typical feature of osteogenesis imperfecta is hearing loss that occurs in childhood, then rapidly progresses. The incidence of deafness in childhood is between 10 and 20%; by middle age, approximately 50% of patients suffering from osteogenesis imperfecta will also be suffering from deafness. The hearing loss may increase during pregnancy. The audiometric pattern of hearing loss is indistinguishable from that of otosclerosis. Typically, the hearing loss commences at puberty, when fractures become less frequent. A conductive hearing loss is present in nearly 80% of cases. Very often a mixed hearing loss is present. On occasion, a purely sensorineural hearing loss may be present, and on rare occasions profound irreversible sensorineural hearing loss is present (Pedersen 1985; Shapiro et al 1982; Stewart and O’Reilly 1989). Sensorineural hearing loss in OIT is believed to be due to encroachment of reparative vascular and fibrous tissue in and about the cochlea or as the result of microfractures and/or hemorrhages (Shapiro et al 1982). Several studies have indicated that the incidence of hearing loss of both types increases with age, as does the proportion of patients with mixed types (Garretsen et al 1997). In one study (Stewart and O’Reilly 1989), 11 out of 12 patients between ages 40 and 49 were affected, as were all 7 patients between 50 and 55 years of age. Tympanometry shows increased compliance values. Stapes fixation is almost always associated with a conductive hearing loss. Hypermobility of the tympanic membrane can be seen simultaneously with fractures or aplasia of the crura of the stapes. Hypermobility of the tympanic membrane occurs because the tympanic membrane has the same embryological origin as the sclera. In those patients who have fractures of the stapedial arch, high compliance values are seen on tympanometry. Carhart’s notches are not present, nor is Schwartze’s sign. Usually the conductive hearing losses are not bilateral, but if and when bilateral conductive hearing losses are present, they are not symmetrical. Vestibular symptoms are rare in osteogenesis imperfecta (Shea and Postma 1982). Johnsson and colleagues (1982), however, have described extensive bilateral endolymphatic hydrops in a patient who suffered from osteogenesis imperfecta. Deposition of osteopenic immature bony tissue that is weak is characteristic of osteogenesis imperfecta. Often, microfractures can be found. Histologically, the number of osteocytes is increased in both woven and lamellar bone accompanied by a relative reduction of matrix substance. The bone turnover rate is very high. OIT has multiorgan manifestations such as fragile bones, hearing loss, dentinogenesis imperfecta, loose joints, mitral valve prolapse, easy bruising, and growth deficiency (Bergstrom 1977; Marini 1988). Although there are some similarities between otosclerosis and osteogenesis imperfecta, both are separate and distinct entities, with defects in the stapes as seen in osteogenesis imperfecta being quite different from those seen in otosclerosis (Berger et al 1986; Pedersen et al 1985). Biochemical analysis of stapes sulfhydryl groups and various enzymes have clearly demonstrated different concentrations in both of these diseases (Arslan and Ricci 1963; Chevance 1964; Holdsworth et al 1973). In most cases, stapedial fixation is caused by a focal lesion in the footplate that histologically resembles the early active stage of otosclerosis. On occasion, fixation of the stapedial foot-plate is the result of diffuse structural alteration of the entire footplate. The extensive degree of disorganization is more pronounced in osteogenesis imperfecta than in otosclerosis (Brosnan et al 1977). Biochemical assays of serum calcium, phosphorus, and calciferol are normal. Alkaline phosphatase levels occasionally may be elevated. Photon absorptiometry has demonstrated that the cortical thickness of bone is reduced; this is not seen in otosclerosis. Temporal bone findings in OIT are nearly identical to those in otosclerosis (Table 17–2). Diffuse resorption of vast areas of the otic capsule is commonly seen in OIT. In advanced cases of OIT, the middle ear cleft can become narrowed by proliferative bone, and the oval and round windows can be obliterated by the dysplastic process. At this time, there is no known cure for OIT. Treatment is instead directed at rehabilitation, although medical treatment for OIT remains elusive. Calcitonin, sodium fluoride, and vitamin D have each been found to have poor efficacy. Results of stapedectomy are generally satisfactory (Armstrong 1984) and are similar to those of otosclerosis (Garretsen and Cremers 1990). Stapedectomy, however, should be delayed until well into adulthood (Pederson and Elbrond 1983). By then it is expected that fractures in response to trivial trauma will have ceased (Kosoy and Maddox 1971). The findings of the middle ear include a very thick and soft footplate. The middle ear mucosa appears to be more vascular than normal. There is a high chance of floating footplate. The long process of the incus may be fragile and prone to fracture during crimping. Other findings include thin canal wall skin, brittle scutum, and fragile stapedial crura. Paget’s disease is characterized by excessive remodeling of bone. This disorder typically affects the axial skeleton. Pathognomonic features of Paget’s are spreading osteolytic and osteoblastic changes that commonly affect the pelvis, lumbar spine, skull, femur, and tibia. The typical patient suffering from Paget’s disease has an enlarged skull, progressive kyphosis, bowed legs, and short stature. Males are four times more likely to be affected than females. The geographic distribution of Paget’s disease includes a large concentration of patients in the United Kingdom, Australia, and New Zealand (Detheridge et al 1982). Paget’s disease can occur sporadically, but in 15% of cases there is an auto-somal dominant inheritance pattern (Avioli 1987). Very rarely does the onset of this disease occur before the age of 40; it is commonly encountered after the age of 55. The overall incidence is 3% but increases by the eighth decade (Freeman 1988). Alternating waves of osteoclastic and osteoblastic activity result in haphazard bony resorption followed by deposition of structurally weakened demineralized bone. In the early phase of the disease, bone resorption is accelerated and can be seen radiologically as a lytic area. The marrow then gets filled with fibrovascular tissue that later becomes sclerotic. The sclerosis contains a mosaic pattern of coarse fibers that are composed of irregularly arranged units of bone (Nager 1975). The pelvis is the site that is most commonly involved, followed by the skull. The superficial temporal artery becomes hypertrophied and tortuous. Bone pain is a common symptom. Expansion of bone in and around the foramina can lead to neurologic deficits that can result in manifestations such as hemifacial spasm, trigeminal neuralgia, and optic atrophy (Clarke and Harrison 1978). Spinal cord lesions with vertebral involvement, nerve compression, and vascular steal syndrome are seen with Paget’s disease. Ataxia, quadriparesis, hydrocephalus, and lower cranial nerve deficits can also occur (Davies 1968).
Otodystrophies
OSTEOGENESIS IMPERFECTA
Incidence of Osteogenesis Imperfecta
Signs and Symptoms of Osteogenesis Imperfecta
Otological Features of Osteogenesis Imperfecta
Histology of Osteogenesis Imperfecta
Radiological Findings of Osteogenesis Imperfecta
Treatment of Osteogenesis Imperfecta
Role of Stapedectomy in Osteogenesis Imperfecta
PAGET’S DISEASE (OSTEITIS DEFORMANS)
Incidence of Paget’s Disease
Histology of Paget’s Disease
Nonotologic Features of Paget’s Disease
Otologic Features of Paget’s Disease

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


