Pediatric Ophthalmology
Edited by P. F. Gallin
Thieme Medical Publishers, Inc.
New York ©2000

23

Orbital Diseases
The identification of an acute orbital process in a child produces an understandably high level of anxiety in the parents, referring pediatrician, and child. To maximize the amount of clinical information that is derived from the initial examination, it is important to begin the interaction by alleviating anxiety. In a relaxed setting, a great deal of information can be derived by simply observing a child from across the examination room. Important diagnostic clues include the discomfort of a child, the motility of the lids and globe, the presence of tearing or inflammation, or a focal mass effect in the lid and proptosis.
As the examination continues the history is derived from the child’s caregiver. It should be detailed, containing a review of familial diseases, birth history, trauma, and medical history. The history of the current illness should be carefully taken as details of the pace of the disease will often provide insight into the disease.
Generally, diseases will evolve by one of three patterns. Rapidly evolving proptosis over the course of hours to days generally represents an infectious/inflammatory process or highly malignant lesion. Slowly evolving processes that grow over the course of months to years are generally benign. Proptosis with growth rates in between are a mixed array of lesions including fungal infections and low-grade malignancies. One of the more helpful examinations to determine the time course of the proptosis is a review of old full-face photographs.
The clinical examination should include an assessment of optic nerve function, including visual acuity, color vision, pupil response, and if the child is able a visual field examination. Proptosis can be estimated grossly by examining the child’s globes from the submental view. More accurate measurements can be made with either a Leudde or a Hertel exophthalmometer. The presence of resistance to retropulsion and associated pain will provide information regarding the nature of the orbital disease. The displacement of the globe out of the axial plane gives an indication of the location of the lesion within the orbit.
Motility of the globe and the eyelids is measured as accurately as possible as is the sensory function in the trigeminal distribution. The localization of the lesion within the orbital apex, optic canal, and cavernous sinus is most easily made based on the performance of the sensory and motor nerves within these spaces.
The conjunctiva should be examined for chemosis (a sensitive measure of congestive ouflow obstruction), hemorrhage, and pigmented lesions. Iris nodules and pigment as well as cataract should be identified. The fundus should be examined for papilledema, venous or arterial dilation, and retinal or choroidal lesions.
The most common lesions to affect children are grouped by age in the following sections. The diseases are listed by incidence.
 Birth
Birth
The disease entities that are apparent shortly after birth are virtually limited to congenital conditions. The lesions feature most commonly developmental anomalies and vascular hamartomas (Table 23-1).
| Capillary hemangioma |
| Lipodermoid |
| Lacrimal mucocele |
| Hematic cyst |
| Encephalocele |
| Microphthalmos with cyst |
| Teratoma |
Capillary Hemangioma
Capillary hemangiomas of childhood represent the most common benign orbital tumor in children. The lesions may be superficial to the orbital septum, deep within the orbit without apparent cutaneous involvement, or, in a minority, traverse both the intra- and extraorbital spaces (Fig. 23-1). The cutaneous tumors generally appear within the first month afterbirth. Typically a flat or only slightly elevated lesion is present at first. The lesions tend to grow rapidly in either a continuously progressive fashion or episodically in association with the child’s growth pattern. Growth of the lesion can be anticipated for approximately 1 year, after which involution occurs for up to 4 or 5 years. Children should be monitored during the growth phase to assure that the growth of the tumor does not produce amblyopia (through obstruction of the visual axis or induced astigmatism) or severe cosmetic deformity.
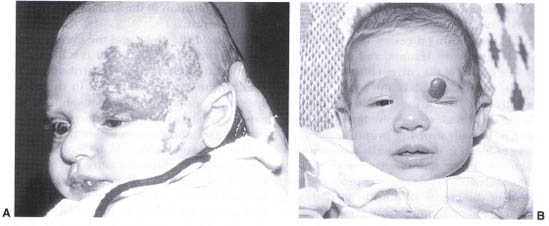
FIGURE 23-1. (A) Capillary hemangioma of the left brow and upper eyelid. (B) Capillary hemangioma of the left upper lid producing significant mechanical ptosis.
Orbital hemangiomas can be more difficult to diagnose. Proptosis is the typical presenting feature and is generally identified by 1 month of age. There may be other cutaneous hemangiomas that suggest the orbital diagnosis. At Babies Hospital we have identified prematurity as a risk factor for the development of capillary hemangioma. Computed tomography (CT) or magnetic resonance imaging (MRI) will identify a noncircumscribed soft-tissue mass. Bone destruction, which is a typical feature of malignancy, is not identified in the case of capillary hemangioma. In some cases a biopsy is required to confirm the clinical suspicion and exclude the diagnosis of malignancy.
As in the case of the cutaneous lesions growth is anticipated during the first year, after which, involution occurs. Patients are monitored for the development of amblyopia and compressive optic neuropathy.
Lesions that extend from cutaneous to the deep orbit have radiographic and clinical features of both processes. The diagnosis is more apparent due to the cutaneous component; however, management may be difficult due to the diffuse involvement of the orbital and adnexal tissues.
Management of this group of lesions is guided by the development of ocular function and cosmetic deformity produced by the lesion. If the lesion is small and slow growing and does not retard visual development, conservative measures are indicated.
Observation at regular intervals will assure that the lesion continues to behave in a benign fashion. After maximal spontaneous resolution, surgical resection of the residual lesion can be considered.
Cutaneous lesions that obstruct the visual axis, producing amblyopia, astigmatism, or significant deformity are treated. Treatment varies with the patient; however, most patients receive corticosteroids by either local injection or systemic administration. Delivered locally, the dose is 40 to 80 mg of triamcinolone and 25 mg of methylprednisolone. If systemic therapy is chosen the dose of prednisone is 1.5 to 2.5 mg/kg daily with the dose and taper guided by the clinical response. The advantage of local therapy is the lower level of systemic absorption and adverse effect. Despite this patients require monitoring by a pediatrician for adrenal axis insufficiency and immunosupression during treatment. The risks of local therapy include atrophy of the overlying skin and the rare but catastrophic blindness resulting from ophthalmic artery occlusion due to propagation of the depo compound during the injection. To avoid injecting into the orbit, deep lesions are uniformly treated with systemic corticosteroids. Alternative therapy has included low-dose radiotherapy, which we presently do not routinely advocate because of the potential for long-term ill effects. Interferon has enjoyed some advocacy; however, it is routinely not indicated due to the need for long-term daily injections of the very costly compound.
When full spontaneous resolution of the orbital hemangiomas has occurred consideration of surgery is given in cases in which proptosis persists. Preoperative noninvasive imaging provides the anatomic details required to plan an effective resection. Complete resection is not routinely possible or required.
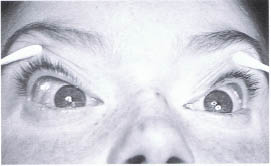
FIGURE 23-2. Lipodermoid of the right superior temporal cul-de-sac.
Lipodermoid
Lipodermoids are relatively common tumors identified in the superotemporal quadrant of the anterior orbit consisting of fatty tissue (Fig. 23-2). The process is most commonly bilateral. The overlying conjunctiva routinely demonstrates hair follicles and aqueous and sebaceous glands. All three may produce ocular irritation and result in a clinical picture of conjunctivitis. The mass is mobile and does not impair ocular motility. The lateral canthal angle may be distorted due to associated maldevelopment. CT scan demonstrates a fat density lesion that may extend deeply into the orbit. The diagnosis of the lesion is made on clinical grounds. Excision of the lesion is contemplated to relieve ocular discomfort and for cosmesis. It should be remembered that no attempt should be made at complete resection as this risks damage to the lateral rectus muscle. Instead a conservative removal of the superficial component of the lesion will achieve the surgical goals.
Lacrimal Sac Mucocele
Lacrimal sac mucoceles result from congenital obstruction of both the valve of Hasner distally and the valve of Rosenm$uUller proximally. This results in a closed lacrimal sac that accumulates mucoid product, resulting in a firm bluish mass generally identified within the first week after birth (Fig. 23-3). These lesions are located beneath the medial canthal tendon and may displace the tendon and attached lower eyelid superiorly. The size of the mass is limited by the distensibility of the surrounding tissues, which is reached within 1 to 2 days. Lacrimal sac mucoceles must be distinguished from deep capillary hemangiomas that generally do not enlarge as rapidly in the first few days of life and encephaloceles that arise above the medial canthal tendon and are not bluish. Further evidence is provided by transillumination, which reveals the cystic nature of the mucocele. If there remains any uncertainty regarding the diagnosis, a CT scan is of value to exclude an encephalocele. Treatment of the mucocele by lacrimal probing is indicated at the earliest convenience to prevent progression to acute dacryocystitis, which often ensues within weeks after birth in these cases.
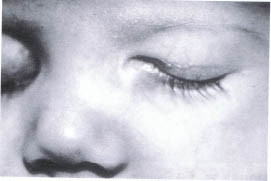
FIGURE 23-3. Mucocele of left nasolacrimal sac. Blue tinged discoloration of the skin is typical of this lesion.
Hematic Cyst
Hematic cysts are an unusual cause of unilateral proptosis identified shortly after birth. In our experience they have been more common among children born prematurely. It is likely that birth trauma is in part responsible for the production of spontaneous bleeding. Although it may be speculated that the hemorrhaging occurs in vascular hamartomas (i.e., lymphangioma, venous malformation), this has not been substantiated histologically. The degree of proptosis can vary from mild to moderate but is not progressive, which helps to distinguish it from a malignancy. The child appears to be in no distress. Ocular function is rarely impaired. MRI identifies the blood-filled cyst (Fig. 23-4). The lesion may be difficult to distinguish from an orbital capillary hemangioma. If there is evidence of compressive optic neuropathy or slowly progressive growth, a trial of oral corticosteroids administered with the aid of a pediatrician or endocrinologist may be helpful. Left untreated the cysts will resolve spontaneously over 1 to 4 weeks. We have not needed to evacuate any of the cases that we have examined.
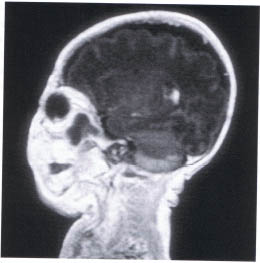
FIGURE 23-4. MRI hematic cyst of the posterior orbit
Encephalocele
A congenital encephalocele occurs as the result of a defect in the closure of a cranial suture that is routinely associated with a craniofacial cleft (Fig. 23-5). The cleft locations have been classified by Tessier. The clefts most commonly associated with orbital encephaloceles are the number 0, 1, 2, 3, 12, 13, and 14 clefts. Clinically the encephalocele is identified by a soft, subcutaneous nontender mass. There may be palpable pulsations coinciding with the natural pulsations of the brain. The mass does not transilluminate. If the encephalocele is located beneath the eyebrow, there may be an associated alopecia. Telecanthus is associated with midline clefts.
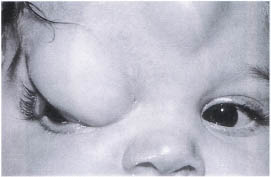
FIGURE 23-5. Encephalocele of the right superior nasal orbit.
Radiographic analysis should be performed by CT scan with particular attention to both bone and soft tissue windows that will identify the cranial bone defect and the prolapsing brain tissue.
Treatment is by craniotomy and orbital bone reconstruction with split cranial bone graft. If telecanthus exists this may be repaired at the same setting.
Microphthalmos with Cyst
At birth the microphthalmic globe is apparent. The malformation may exist in association with a cyst produced at the fourth week of embryology (Fig. 23-6). The cyst will grow slowly, which helps to distinguish it from teratomas. Imaging with ultrasound will identify the cystic nature of the lesion. CT will exclude encephalocele by imaging of the orbital bones. MRI is helpful when examining the soft tissue contents of the cyst. Treatment is surgical excision of the cyst. The globe should be removed if rudimentary and replaced with a dermis fat graft to promote orbital bone and soft-tissue development.
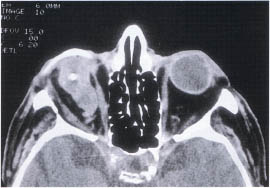
FIGURE 23-6. Microphthalmos with cyst of the left orbit; an internal calcification is noted.
Teratoma
Teratomas are defined as a tumor composed of tissues derived from all three germ cell layers. They are rare, rapidly growing lesions that may compromise normal ocular function (Fig. 23-7A). Radiographic analysis may benefit from both CT (Fig. 23-7B), which will identify the location of the lesion, and any associated bone defects and MRI, which best demonstrates the complexity of the soft-tissue structure of the lesion. If the diagnosis is not certain based on clinical and radiographic grounds, a biopsy is helpful (Fig. 23-7C). Complete resection of the lesion may be possible while preserving normal ocular function.
 Three Months to 1 Year
Three Months to 1 Year
Disease entities that manifest in children between 3 months and 1 year of age are listed in Table 23-2.
| Dermoid cyst |
| Arteriovenous malformation |
| Rhabdomyosarcoma |
| Neuroblastoma |
Dermoid Cysts
Dermoid cysts are the result of ectodermal inclusions at the site of bony suture closure. Any of the orbital or periorbital sutures may be involved (Fig. 23-8A). The lesions arising from the anterior orbital sutures are generally identified within the first several months of birth. Typically the cysts arising from the deep orbital sutures present with proptosis or optic neuropathy in later decades. We have, however, seen two cases in which dermoid cysts of the sphenozygomatic suture produced proptosis in one child at 5 months and the second at 2 years of age. The most common location for the lesions are at the lateral brow (frontozygomatic suture) and the superonasal notch (frontonasal suture). We generally perform CT scans on the lesions in the superonasal quadrant to exclude the possibility of encephalocele or bone dysgenesis preoperatively (Fig. 23–8B). Excision is recommended after 6 months of age to limit anesthetic risk. Removal is advisable to eliminate the risk of rupture of the cyst, which may mimic infectious cellulitis and limit the size of the postoperative scar. To further limit the scar we have to be able to remove most anterior lesions through lid crease incisions.
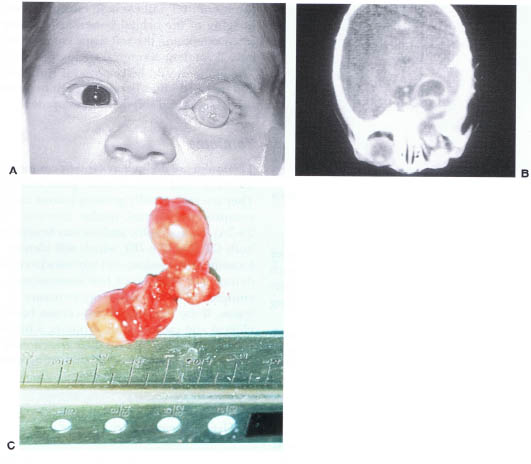
FIGURE 23-7. (A) Teratoma of the left orbit. (B) CT scan of an orbital teratoma demonstrating extension of the lesion into the intracranial space. (C) Surgical specimen of orbitocranial teratoma. (Courtesy of J.A. Katowitz, M.D.)
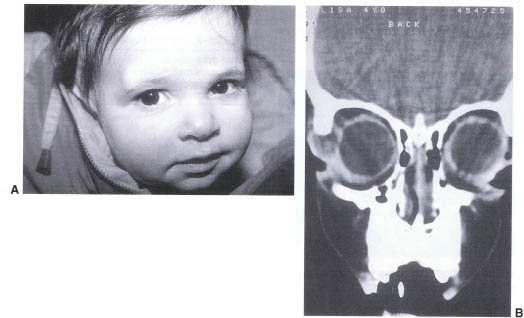
FIGURE 23-8. (A) Dermoid of right superior-temporal orbit. (B) CT scan of a right orbital dermoid demonstrating the cystic lesion of the right frontozygomatic suture.
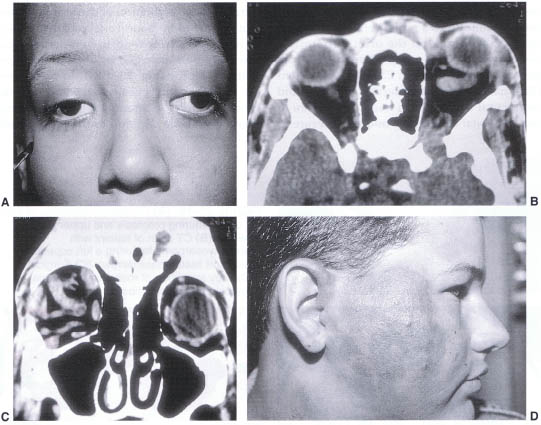
FIGURE 23-9. (A) Child with Osier-Weber-Rendu syndrome. (B) CT scan in axial projection of the same patient demonstrating a dilated superior ophthalmic vein of the left orbit. (C) Coronal CT scan demonstrating a tortuous arteriovenous malformation within the right orbit. (D) Subcutaneous arteriovenous malformation of the right face.
Congenital Arteriovenous Malformation
Arteriovenous (AV) malformations may be of two varieties. The high flow lesions result most commonly from trauma or age-related hypertensive shunting. The congenital lesions are more often of a lower flow rate and grow slowly over decades. Unlike capillary hemangiomas, AV malformations tend to be identified several months after birth and continue to grow slowly past 1 year (Fig. 23–9D), the point at which hemangiomas will begin to involute. The AV malformations may occur as part of the Wyburn-Mason or Osler-Weber-Rendu syndromes (Fig. 23–9A–C). Examination for visceral or retinal involvement should be undertaken. In general, the lesions should be left untreated unless there is significant visual compromise or cosmetic deformity. In such cases angiography to identify discrete arterial feeding vessels that can be occluded intravascularly may be curative. Often the lesion will arborize new vessels that will require further treatment. Surgical resection is indicated to remove devitalized tissue that produces significant residual volume after arterial occlusion or in cases of vision-threatening orbital inflammatory response to the intravascular thrombotic agent.
Rhabdomyosarcoma
Rhabdomyosarcoma is the most common orbital malignancy in the pediatric population. Over 70% of rhabdomyosarcomas are identified by the end of the first decade of life, with a high frequency within the first year. Most are identified within the first 2 decades, but occasional lesions are noted in later decades. The lesion presents clinically as a rapidly enlarging mass if superficially located in the orbit. Proptosis is common if the lesion is present behind the equator of the globe (Fig. 23–10A). Associated inflammatory signs including skin erythema and conjunctival chemosis can mimic those seen in cases of orbital cellulitis or trauma. It is rare to identify a case in which the growth rate is slow. In such cases the diagnosis may be delayed.
The evaluation of a child with a rapidly enlarging orbital mass should include a CT scan with contrast to locate the lesion, define its anatomic extension, and identify its effect on the surrounding orbital bones (Fig. 23-10B). MRI provides added detail of the soft-tissue invasion but does not improve on the diagnostic capabilities of CT. On both CT and MR the lesion is an unencapsulated soft-tissue mass that enhances with contrast injection. The orbital structures are displaced by the mass. Bone destruction is not universal, and extension through the orbital fissures is known to occur.
The clinical radiographic features should alert the physician to the possibility of an orbital malignancy. An orbital exploration should follow almost immediately. A decision regarding the extent of surgery requires careful preoperative evaluation of the imaging studies and an intraoperative evaluation of the lesion. Tumors that appear to be poorly encapsulated on scan or intraoperatively tend to hemorrhage profusely are best managed by obtaining a representative biopsy (Fig. 23-10C). In cases where the lesion is better encapsulated and resection proceeds easily, an attempt at gross total resection is a valuable addition to the postoperative treatment planning. Specimens should be obtained for both formaldehyde and glutaraldehyde fixation and electron microscope evaluation.
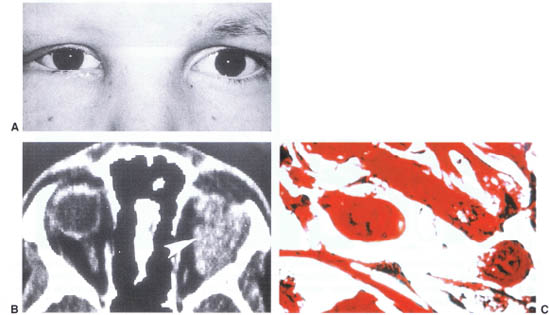
FIGURE 23-10. (A) Rhabdomyosarcoma of the left orbit featuring proptosis and upper eyelid swelling. (B) CT scan of patient with rhabdomyosarcoma featuring a left superior orbital soft tissue mass (arrow). (C) Surgical pathology specimen of rhabdomyosarcoma demonstrating muscle fibers.
There are four histopathologic subtypes of rhabdomyosarcoma: embryonal, alveolar, pleomorphic, and botryoid. The most common form is embryonal (65% of tumors). Alveolar is poorly differentiated and accounts for 30% of cases. Pleomorphic is most common in adults and is identified in only 1 to 2% of cases. It is unclear whether the histologic type of tumor bears on the survival rate. However wide tumor extension and the presence of parameningeal disease both adversely affect survival.
Prior to 1972 survival rates in cases treated with only wide local excision were only in the 20% range. Since the advent of combination therapy with radiation and chemotherapy the survival rate has increased to nearly 90%. Current protocols developed by the rhabdomyosarcoma intergroup study include multiple regimens of chemotherapy with and without adjunct radiotherapy. The final conclusions of the study will hopefully allow for reduced morbidity while preserving the high survival rate associated with the disease.
Recurrent local disease is a more difficult problem. Current recommendations include wide local excision of the involved tissue and surrounding bone and sinuses where required. This is followed by chemotherapy and, if not previously employed, radiotherapy. Long-term survival in such cases is less favorable.
The long-term effects of the treatment include orbital hypoplasia, retinopathy, optic neuropathy, and cataract. Second malignancies, frequently leukemia, are also seen.
Neuroblastoma
Neuroblastoma is the most frequently occurring metastatic tumor of childhood. Although most are identified prior to 2 years of age the rate of treatment success declines precipitously after 1 year. The classic presentation is the development of rapidly progressive proptosis with associated periorbital ecchymosis (Fig. 23-11A). Limitation of ocular rotations follows soon after with chemosis and finally compressive optic neuropathy. There may be fullness of the temporalis fossa when the lesion erodes through the sphenozygomatic suture (Fig. 23-11B). Bilateral metastatic disease has been reported in fewer than 50% of cases. Intraocular metastatic lesions are rare. A clinical examination should include abdominal palpation to inspect for the primary adrenal or paraspinal mass.
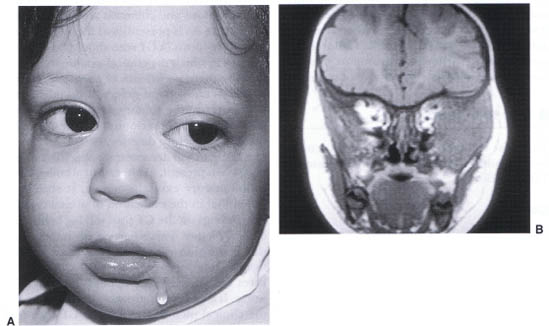
FIGURE 23-11. (A) Child with left orbital neuroblastoma. (B) MRI of neuroblastoma featuring soft tissue mass of the left lateral orbit extending into the temporalis fossa and down into the infratemporal fossa.
CT scan with contrast enhancement shows an enhancing lesion without encapsulation that frequently produces surrounding lytic bone destruction. The site of the metastatic lesion is most frequently the bone marrow space of the sphenoid bone. Extension into the middle cranial fossa is common. If the lesion on CT confirms the clinical suspicion for neuroblastoma, a CT of the chest and abdomen should follow. Further systemic evaluation should include blood and urine evaluation for elevated catecholamines and their metabolites.
A confirmatory diagnostic biopsy should be coordinated with a pediatric oncologist and pediatric surgeon. At the time of the orbital biopsy a bone marrow biopsy and placement of a central venous catheter can be accomplished at the same operative setting and permit rapid institution of therapy. The orbital biopsy should be performed through the safest and most direct route. Debulking the tumor is of little value and may result in damage to the surrounding normal structures.
Treatment varies among institutions but invariably involves chemotherapy (including cisplatin, cyclophosphamide, vincristine, and adriamycin). Treatment response is evaluated at 4 to 6 months and the residual tumor surgically resected. Resistant tumors have been treated with total body irradiation or high-dose chemotherapy followed by bone marrow transplantation. Success is best achieved in children less than 1 year of age (72%). After 2 years of age the survival rate falls to 12%. Children presenting with Horner’s syndrome or opsoclonous appear to have a higher rate of success.
 One to 6 Years
One to 6 Years
The most common ocular diseases that manifest in children aged 1 to 6 years are listed in Table 23-3.
Cellulitis
Infectious orbital cellulitis is the most common cause of proptosis in the pediatric population. The most common source of infection is by contiguous spread from the paranasal sinuses, although less common sources are hematogenously delivered to the orbit or by transcutaneous puncture wounds. As such infectious orbital cellulitis generally becomes a clinically significant entity after the first year of life, when the paraorbital sinuses begin to pneumatize. The ethmoid sinuses are the first to develop followed by the maxillary sinuses within the first 2 years after birth. The frontal sinus develops between the fifth and seventh year, and the sphenoid sinuses are the last to pneumatize in the second decade.
| Cellulitis |
| Dacryoadenitis |
| Lymphangioma |
| Neurofibroma |
| Schwannoma |
| Histiocytosis X |
| Optic glioma |
| Optic meningioma |
| Leukemia/lymphoma |
The typical clinical picture is one of a child who has suffered an upper respiratory tract infection and develops unilateral or occasionally bilateral lid swelling (Fig. 23-12). This is followed within 1 or 2 days by proptosis and complete closure of the lids. The child is generally febrile and lethargic. Vision is often difficult to measure in these cases, but pupil reactions may be assessed to monitor for a relative afferent pupil defect. Ocular motility may be impaired if there is a subperiosteal collection. Proptosis and chemosis are often present. A leukocytosis is present, and there is a shift to immature forms. A CT scan should be obtained in cases in which the clinical examination is difficult or there is evidence of proptosis, motility impairment, or pupil defect. If lid edema and erythema are the solitary signs, medical treatment with close observation is sufficient unless the clinical picture deteriorates.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


