Fig. 1
Photo of arhinia
Initial management of arhinia may require tracheotomy to provide a safe airway with deferred surgical planning as a child or adolescent once the degree of cognitive development has been established. For high-functioning children, a craniofacial team approach of otolaryngology, plastic surgery, and potentially neurosurgery is required to establish nasal patency followed by external nasal reconstruction. Both CT and MRI may be complementary for surgical planning.
Anterior (Pyriform) Stenosis
Congenital nasal pyriform aperture stenosis (CNPAS) is an unusual narrowing of the anterior nasal vault. Anatomically, the anterior nasal vault is defined by the nasal bones superiorly, the nasal process of the maxilla laterally and the junction of the horizontal process of the maxilla and the anterior nasal spine inferiorly.
This entity was first described in adults in 1952 [10] and in a neonate in 1989 [11]. CNPAS may be an isolated anomaly or associated with other craniofacial malformations. The most common is the holoprosencephaly spectrum including a single central upper incisor tooth, absent upper labial frenulum, and absence of the corpus callosum [12, 13]. In mild cases, the neonate may have stertor and mild difficulty coordinating breathing and feeding. In more severe cases, the child may have symptoms and signs that mimic atresia with cyclical cyanosis and respiratory distress. Often, patients will pass the mirror test since there is stenosis but not atresia. Flexible nasal endoscopy may demonstrate the inability to pass the infant fiberoptic telescope. CT scan is diagnostic and helps to rule out other midline anatomic abnormalities. Beldon performed CT analysis and determined that the normal term infant should have at least an 11 mm width across the pyriform aperture Fig. 2 [14].
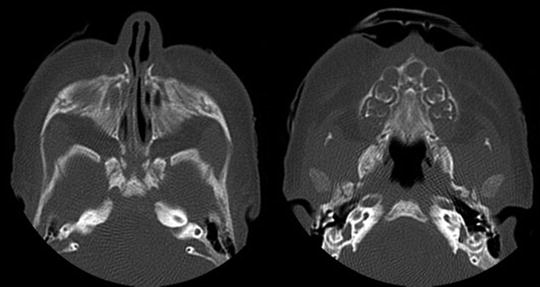
Fig. 2
CT scan of pyriform stenosis and single central incisor associated with holoprosencephaly spectrum
Initial management depends on the degree of respiratory distress. In isolated instances, intubation is rarely necessary. However, in syndromic neonates, establishment of a safe airway may be necessary. If multiple airway anomalies are present, nasal surgery alone may not be sufficient and a tracheotomy is required. When the lesion is isolated, surgery with nasal stenting is very successful.
Mid-vault Stenosis
Rarely, a congenital thickening of the nasal septum or narrowing of the total vault may cause mid-vault stenosis premature infants. Typically, this is self-limited and responds well to topical steroid treatment.
Posterior (Choanal) Stenosis or Atresia
Choanal atresia (CA) is a relatively rare malformation occurring in approximately 1 in 7,000–8,000 births. Unilateral atresia is more common than bilateral. Previous textbooks report that CA is more common in females than males; however, more recent reviews report an equal gender distribution [15, 16]. The purported mechanism leading to CA is either failure of buccopharyngeal membrane breakdown or persistence and misdirection of mesoderm during embryogenesis. In only rare circumstances is the obstruction due to soft tissue alone. In almost all cases, there is some bony component that needs to be removed for successful surgical treatment.
Bilateral CA is diagnosed shortly after birth due to characteristic cyclical cyanosis. The mirror test or inability to pass a 5–6 Fr catheter is suspicious for CA. Flexible fiberoptic endoscopy is diagnostic in most cases Fig. 3. Occasionally, an obstructing posterior nasal or nasopharyngeal growth may mimic CA. Fine cut CT scan is essential to determine the site and degree of obstruction, and image the presence of the nasopharyngeal roof and cavity. Unilateral CA may go unrecognized for years and often presents during childhood or adolescence as inability to breathe through one side of the nose with constant unilateral runny nose. Likewise, choanal stenosis as opposed to atresia may go unrecognized for years.
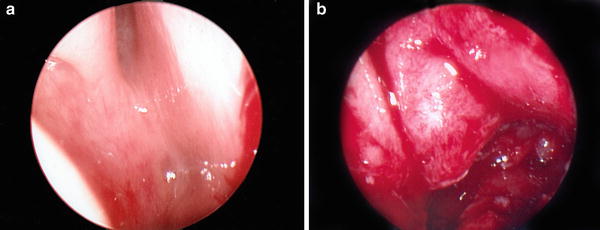
Fig. 3
(a) Endoscopic photo of choanal atresia preop. (b) Endoscopic photo of choanal atresia postop
Every neonate with choanal atresia should be screened for CHARGE syndrome. CHARGE used to be classified as an association. However, with the discovery of the genetic marker CHD7 on Chromosome 8, which encodes the chromodomain helicase DNA binding protein, CHARGE is now classified as a syndrome [17]. CHARGE syndrome is characterized by ocular Colobomas, Heart defects, choanal Atresia, Retarded growth, Genitourinary hypoplasia, and Ear abnormalities. CHD7 analysis in CHARGE syndrome detects mutations in 65–70 % of individuals. Cardiovascular abnormalities occur in 75–85 % and tracheoesophageal fistula is present in 15–20 % of newborns with CHARGE syndrome. Ossicular malformations, cochlear anomalies, and semicircular canal hypoplasia occur in over 80 % of patients [17].
Bilateral CA is a medical urgency; however, it is not a surgical emergency. Use of an oral prop for adequate breathing and an orogastric feeding tube for nutrition allows for the orderly evaluation of other potential anomalies, proper counseling, and surgical planning. With the advent of endoscopic equipment, miniaturization of power-assisted drills and shavers, surgical repair of choanal atresia has evolved over the past 20 years. Whereas previous textbooks discuss both transpalatal and transnasal approaches, the vast majority of current repairs are performed using transnasal techniques. Whether or not to place a short-term stent is still debated in the literature [18–21]. The most difficult cases involve syndromes with severe down sloping nasopharyngeal roofs or nearly absent nasopharyngeal cavities such as Treacher Collins syndrome.
Congenital Masses
Nasolacrimal Duct Cyst (Dacryocystocele)
Obstruction of the nasolacrimal duct is a common anomaly with widely varying reported rates [22, 23]. Many duct obstructions are partial and self-correct by 1 year of age. The duct is usually obstructed at the valve of Hasner just lateral to the inferior turbinate [24]. This may lead to a round mass in the lower nasal cavity. If the proximal valve is obstructed at the common canniculus, this leads to a round facial swelling inferior to the medial canthus of the eye. This may become infected with impressive swelling and erythema mimicking acute unilateral sinusitis. Occasionally, when bitateral cysts are present, this may lead to severe nasal obstruction in the neonate and difficulty coordinating breathing and feeding similar to bilateral choanal atresia. When the clinical picture is uncertain, CT scan with contrast or MRI helps to distinguish between bilateral nasolacrimal duct obstruction, anterior pyriform stenosis, or other mass effects due to tumor.
Surgical treatment and timing varies depending on surgeon preference and severity of presentation. Unilateral dacryocystoceles rarely need urgent surgical correction. However, bilateral obstruction may require surgery within the first few weeks of life. Treatment ranges in escalation from duct probing and balloon dilation for simple duct obstruction to intranasal marsupialization and silastic stenting for large obstructing cysts [25].
Infantile Hemangioma
Infantile hemangiomas are the most common benign neoplasms of infancy. The most common site is in the head and neck primarily in the central facial region. Occasionally, a large nasal and facial hemangioma may proliferate quickly in the intranasal region leading to obstructed breathing. The traditional treatment for many years had been intralesional injection of steroids or systemic steroids with laser ablation during the proliferative phase. However, in recent years, propranolol treatment has supplanted older modalities [26, 27]. Propranolol has been identified on molecular analysis to block endothelial cell proliferation, migration, and formation of the actin cytoskeleton with alteration in vascular endothelial growth factor receptor- 2 [28]. Neonates and infants should be referred to a center familiar for multidisciplinary evaluation and management [29].
Meningocele, Meningoencephalocele, and Glioma (Heterotopic Glial Tissue)
Meningoceles, meningoencephaloceles, and gliomas are often grouped together. A meningocele represents downward displacement into the nose of only the meninges due to small defects in the floor of the intracranial cavity. A meningoencephalocele results from a larger defect that allows for herniation of both meninges and brain tissue. This may extend outward at the glabellar region or only downward with a cribiform plate defect with a large intranasal mass that resemble a polyp. By definition, a nasal glioma is sequestered or displaced neural tissue with fibrous and vascular connective tissue that increases the firmness of the mass. There may be a connecting fibrous stalk to the skull base in 10–15 %; however, it is rare to have any meningeal connection. Therefore, clinically, a meningoencephalocele is soft, compressible, pulsatile; whereas a glioma is firm, noncompressible, and non-pulsatile.
CT scan helps to identify the bony defect in the skull base. MRI is much more useful to demonstrate the characteristics of the soft tissue mass and locate the site of intracranial extension Fig. 4. On T1-weighted MRI, the intranasal component is isointense or hypointense and on T2-weighted images is hyperintense relative to brain tissue. Gliomas are 60 % extranasal, 30 % intranasal, and 10 % combined (dumbbell growth).
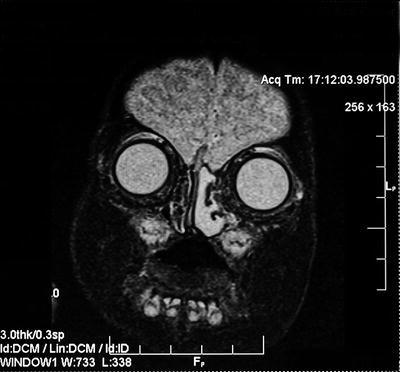
Fig. 4
CT scan of left intranasal glioma: note distinct separation from dura
Surgery on a meningoencephalocele requires a neurosurgeon and otolaryngologist. Surgery is typically via anterior frontal craniotomy [30]. Glioma surgery is typically performed via the intranasal approach and usually does not require intraoperative neurosurgical involvement; however, a neurosurgeon should be available on a standby basis for any unexpected encounter or complication Fig. 5 [31, 32].
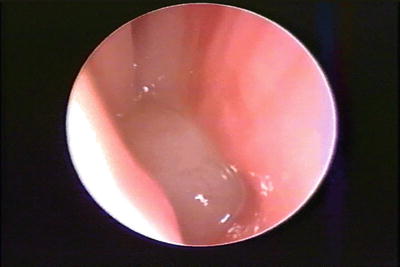
Fig. 5
Endoscopic photo of nasopharyngeal glioma
Nasal Dermoid Sinus and Cyst
Derived from ectoderm and mesoderm, a dermoid cyst or sinus may form due to a remnant in the prenasal space posterior to the nasal bones and anterior to the nasal and septal cartilages. The typical sinus tract has a pit and related small hair tuft on the nasal dorsum. This may present as an infected midline mass or with keratin discharge. The tract is usually well-defined on imaging. However, in some cases, the distal extent may project anteriorly all the way to the nasal tip altering surgical planning or increasing the likelihood of recurrence. CT scan helps to define bony skull-base defects and widening of the crista galli, which is associated with potential intracranial extension. MRI is very useful to determine the extent of the tract and mass, and serpentine extensions and intracranial extension.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


