Purpose
To determine the frequency of ocular adverse effects associated with vemurafenib (PLX4032) treatment for metastatic cutaneous melanoma.
Design
Retrospective review of the clinical study reports from the clinical pharmacology, phase 1, phase 2, and phase 3 trials of vemurafenib.
Methods
The vemurafenib clinical trials were a multicenter series involving adult patients with histologically confirmed, BRAF V600 mutation–positive, unresectable, stage IIIC or IV melanoma. A total of 855 patients were enrolled in the trials: 568 patients were treated with vemurafenib and 287 patients were treated with dacarbazine.
Results
Among the 568 patients treated with vemurafenib, ocular adverse effects developed in 22% (95% confidence interval [CI], 18.5–25.6). The most common ocular diagnosis was uveitis (4.0%; 95% CI, 2.6–6.0), followed by conjunctivitis (2.8%; 95% CI, 1.6–4.5) and dry eyes (2.0%; 95% CI, 1.1–3.7). All were successfully managed while vemurafenib therapy was continued.
Conclusions
Ocular adverse events and symptoms may be seen in more than one-fifth of patients being treated with vemurafenib. However, vemurafenib can be continued while the ocular symptoms are being managed. The pathogenesis of ocular symptoms in this patient population is unclear; additional studies are necessary.
Metastatic cutaneous melanoma has historically been associated with a dismal prognosis. Despite advances in surgical, chemotherapeutic, and radiation techniques, the overall survival rate for metastatic cutaneous melanoma has remained largely unchanged in the past 20 years. Recently, therapeutic targeting of the mitogen-activated protein kinase (MAPK) pathway (the RAS-RAF-MEK–extracellular regulated kinase [ERK] cascade) has shown significant success in improving clinical outcomes in patients with advanced cutaneous melanoma ( Figure 1 ).
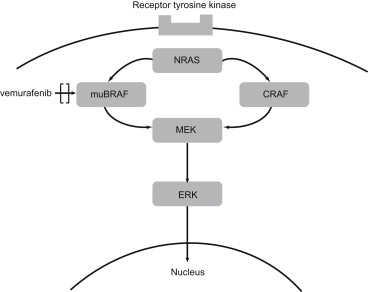
In 2002, Davies and associates made the landmark discovery of activating V600 mutations in BRAF (v-raf murine sarcoma viral oncogene homolog B1) in the MAPK pathway. This mutation most commonly is a single substitution of valine 600 to glutamic acid in the activating region of the kinase domain ( BRAF V600E ). The BRAF V600E and other BRAF V600 mutations are found in 40%–60% of melanomas and result in RAS-independent activation of RAF kinase. This causes constitutive phosphorylation of downstream ERK, which leads to the transduction of intracellular signals that promote cell proliferation and cancer development.
Vemurafenib is a potent and specific oral BRAF V600 inhibitor that has produced more than a 50% objective response rate in phase 1 and 2 trials. In a randomized phase 3 trial comparing vemurafenib and the alkylating chemotherapy agent dacarbazine, vemurafenib had a relative reduction in the risk for death of 63%. This discovery led to agent approval by the US Food and Drug Administration (FDA) and widespread clinical use around the world. The adverse effects most commonly reported with vemurafenib therapy are cutaneous and include rash, alopecia, pruritus, hyperkeratosis, and an increased incidence of keratoacanthoma and squamous cell carcinoma. Other common adverse effects include arthralgia, nausea, fatigue, diarrhea, headache, vomiting, lymphopenia, and neutropenia.
Little has been written in the literature about the ocular adverse effects of vemurafenib. Herein we describe the incidence of ocular adverse effects within the 4 published clinical trials of vemurafenib.
Methods
Clinical study reports from cutaneous melanoma patients treated with therapeutic doses of vemurafenib (>240 mg orally 2 times daily) who were enrolled in any of the following clinical trials—clinical pharmacology, phase 1 (PLX-06-02), phase 2 (BRAF Inhibitor in Melanoma [BRIM-2]), and phase 3 (BRIM-3) studies, together with the 3-month safety update (for the clinical pharmacology, BRIM-2, and BRIM-3 studies only [June 2011])—were used to identify patients who had experienced ocular adverse events during treatment with vemurafenib. For the BRIM-3 study, patients who received the active comparator dacarbazine during the study and who experienced ocular adverse events were also identified. For all patients enrolled in these studies, ocular adverse effects were recorded according to Common Terminology Criteria for Adverse Events (CTCAE), version 3.0. In particular, the patient listings from each report were retrospectively reviewed to identify all relevant patients. Protocols for the studies were approved by the appropriate institutional review boards at the time of the study; conformed to all country, federal, or state laws; and adhered to the tenets of the Declaration of Helsinki. (For the specific review boards for each institution, refer to the reports of the phase 1, 2, and 3 trials. ) All patients signed written informed consent prior to enrollment in the clinical trials. De-identified patient information that conformed to Health Insurance Portability and Accountability Act (HIPAA) guidelines was available for retrospective analysis for this study; all patients enrolled in the vemurafenib clinical trials consented to use of this information. The clinical trials in this manuscript were registered with the National Institutes of Health and can be found on the www.clinicaltrials.gov website with the following identifiers: NCT00405587 , NCT00949702 , NCT01006980 , and NCT01107418 .
Uveitis was the most common ocular adverse event in this study population; therefore, this subgroup was analyzed separately. The following preferred terms were included in the definition of uveitis: uveitis, intermediate uveitis, iritis, iridocyclitis, and vitritis. After patients with uveitis were identified, key baseline characteristics of the study patients were compared and the treatment methods were recorded.
Results
Among 855 patients enrolled in the vemurafenib clinical trials, 568 were treated with vemurafenib and 287 were treated with dacarbazine ( Table 1 ). An ocular adverse effect developed in 125 of the 568 patients (22.0%) who were treated with vemurafenib (95% confidence interval [CI], 18.5–25.6). Among the 287 patients treated with dacarbazine, 10 (3.5%; 95% CI, 1.7–6.3) reported ocular adverse effects ( Table 1 ).
| Trial | CPS | Phase 1 | Phase 2 | Phase 3 | Total | |
|---|---|---|---|---|---|---|
| Drug | Vem (n = 52) | Vem (n = 48) | Vem (n = 132) | Vem (n = 336) | DTIC (n = 287) | Vem (n = 568) |
| Patients with ≥1 ocular adverse effect, n (%) | 3 (5.8) | 25 (52.1) | 35 (26.5) | 62 (18.5) | 10 (3.5) | 125 (22.0) |
| Diagnosis, n (%) | ||||||
| Uveitis | 0 | 7 (14.6) | 7 (5.3) | 9 a (2.7) | 0 | 23 (4.0) |
| Conjunctivitis | 0 | 6 (12.5) | 0 | 10 (3.0) | 1 (0.3) | 16 (2.8) |
| Dry eyes | 1 (1.9) | 4 (8.3) | 1 (0.8) | 6 (1.8) | 0 | 12 (2.1) |
| Episcleritis | 0 | 0 | 3 (2.3) | 0 | 0 | 3 (0.5) |
| Keratitis | 0 | 1 (2.1) | 0 | 1 (0.3) | 0 | 2 (0.4) |
| Blepharitis | 0 | 0 | 0 | 3 (0.9) | 0 | 3 (0.5) |
| Retinal vein occlusion | 0 | 0 | 1 (0.8) | 0 | 0 | 1 (0.2) |
| Other eye disorders b | 0 | 3 (6.25) | 7 (5.3) | 4 (1.2) | 12 (4.2) | 14 (2.7) |
| Symptoms, n (%) | ||||||
| Blurred vision | 0 | 4 (8.3) | 8 (6.1) | 9 (2.7) | 2 (0.7) | 21 (3.7) |
| Ocular discomfort c | 1 (0.3) | 2 (4.2) | 2 (1.5) | 15 (4.5) | 5 (1.7) | 20 (3.5) |
| Photophobia | 0 | 7 (14.6) | 4 (3.0) | 6 (1.8) | 2 (0.7) | 17 (3.0) |
| Ocular injection | 1 (0.3) | 0 | 4 (3.0) | 11 (3.3) | 1 (0.3) | 16 (2.8) |
| Tearing | 0 | 2 (4.2) | 3 (2.3) | 6 (1.8) | 0 | 11 (1.9) |
| Periocular edema | 0 | 0 | 3 (2.3) | 4 (1.2) | 0 | 7 (1.2) |
| Diplopia | 0 | 1 (2.1) | 2 (1.5) | 2 (0.6) | 1 (0.3) | 5 (0.9) |
| Floaters | 0 | 2 (4.2) | 0 | 2 (0.6) | 1 (0.3) | 4 (0.7) |
a Three patients in the phase 3 study experienced 2 occurrences of uveitis; therefore, there were 12 events among a total of 9 patients.
b Other reported ocular adverse events include asthenopia, discharge, ectropion, scleral cyst, mydriasis, retinal pigment epitheliopathy, subconjunctival hemorrhage, cataract, chalazion, scintillating scotoma, corneal lesion, eye pain, photophobia, blurred vision, diplopia, eye irritation, reduced visual acuity, visual impairment, and vitreous floaters.
Of a total of 568 patients, uveitis (4.0%; 95% CI, 2.6–6.0) was the most common ocular diagnosis associated with vemurafenib therapy, occurring in 23 patients, followed by conjunctivitis in 16 patients (2.8%; 95% CI, 1.6–4.5) and dry eyes in 12 patients (2.0%; 95% CI, 1.1–3.7). Patients commonly reported blurred vision, ocular discomfort, photophobia, redness, tearing, and edema—symptoms consistent with ocular inflammation.
Patients With Vemurafenib-Associated Uveitis
Across the 4 clinical trials, 23 patients had uveitis during the study period. The clinical demographics of this patient subset were similar to the demographics of the general population of these clinical trials ( Table 2 ). Mild to moderate uveitis (CTCAE grade 2 or 3) was diagnosed a median of 117 days after initiation of vemurafenib therapy (range, 7–550 days) ( Figure 2 and Table 3 ). No cases of grade 4 uveitis (blindness) were observed. Recurrent uveitis developed in 4 patients during the study period. All patients continued vemurafenib; however, in 6 of the 23 patients (26%), the dose was reduced because of the diagnosis of uveitis. Uveitis was treated successfully in the majority of patients by use of standard local therapies, including topical or periocular corticosteroids, cycloplegic agents, and agents to lower intraocular pressure as needed. In a subset of patients, intermittent systemic or intraocular steroids were used. In all cases of uveitis, it was possible to continue vemurafenib.
| Phase 1 | Phase 2 | Phase 3 | Total | |
|---|---|---|---|---|
| Uveitis (n = 7) | Uveitis (n = 7) | Uveitis (n = 9) a | Uveitis (n = 23) | |
| Median age (range) (y) | 56 (22–68) | 43 (24–72) | 57 (41–76) | 56 (22–76) |
| Male sex, n (%) | 2 (29) | 3 (43) | 7 (78) | 12 (52) |
| ECOG PS, b n (%) | ||||
| 0 | 5 (71) | 6 (86) | 7 (78) | 18 (78) |
| 1 | 2 (29) | 1 (14) | 2 (22) | 5 (28) |
| Stage of metastatic melanoma, c n (%) | ||||
| M1c | 5 (71) | 4 (57) | 7 (78) | 16 (70) |
| M1b | 0 | 0 | 1 (11) | 1 (4) |
| M1a | 2 (29) | 3 (43) | 1 (11) | 6 (26) |
| Unresectable IIIC | 0 | 0 | 0 | 0 |
| Prior therapies, n (%) | ||||
| 0 | 2 (28.6) | 0 | 0 | 2 (8.7) |
| 1 | 3 (42.9) d | 4 (57.1) | n/a | 7 (30.4) |
| 2 | 2 (28.6) | 3 (42.9) | n/a | 5 (21.7) |
| ≥3 | 0 | 0 | n/a | 0 |
| Other vemurafenib-related toxicities, n (%) | ||||
| Rash | 6 (85.7) | 2 (28.6) | 4 (44.4) | 12 (52.2) |
| Arthralgia | 5 (71.4) | 6 (85.7) | 2 (22.2) | 13 (56.5) |
| Fatigue | 4 (57.1) | 4 (57.1) | 5 (55.6) | 13 (56.5) |
| SCC/keratoacanthoma | 4 (57.1) | 1 (14.3) | 3 (33.3) | 8 (34.8) |
a Three patients in the phase 3 study experienced 2 occurrences of uveitis; therefore, there were 12 events among a total of 9 patients.
b Grade 0 = fully active, able to carry on all predisease performance without restriction; grade 1 = restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature (eg, light housework, office work).
c M1a = metastases to skin, subcutaneous, or distant lymph nodes; M1b = metastases to lung; M1c = metastases to all other visceral sites or distant metastases to any site combined with an elevated serum lactate dehydrogenase; IIIC = clinically palpable lymph node, or unresectable locally advanced disease. Staging criteria follow the American Joint Committee on Cancer (AJCC) TNM staging system.
d One patient required 2 consecutive courses of interleukin 2 therapy; however, this was counted as 1 therapy type.



