Drug (or drug candidate)/trade name
Indications
Status
References
Acyclovir
Antiviral drug
Under development
Atropine sulfate
Treatment of peptide ulcer disease
Under development
Behl et al. (1998)
Apomorphine
Medication for erection
Under development
Costantino et al. (2007)
Buprenorphine hydrochloride
Analgesic drug
Under development
Behl et al. (1998)
Butorphanol/Stadol NS®
Migraine
Market
Grassin-Delyle et al. (2012)
Carvedilol
Cardiovascular drug
Under development
Estradiol/Aerodiol®
Hormone replacement therapy
Market
Fentanyl/Instanyl®, PecFent®
Breakthrough pain
Market
Haloperidol
Antipsychotic
Under development
Grassin-Delyle et al. (2012)
Ketamine
Analgesic drug
Under development
Bitter et al. (2011)
Meclizine hydrochloride
Management of nausea, vomiting and dizziness
Under development
Behl et al. (1998)
Metoclopramide hydrochloride
Antiemetic drug
Under development
Morphine
Analgesic drug
Under development
Naloxone/Narcan®
Treatment of opioid overdose
Market
Costantino et al. (2007)
Nicotine/Nicotrol NS®
Smoking cessation
Market
Grassin-Delyle et al. (2012)
Nifedipine
Management of vasospastic angina
Under development
Nitroglycerin
Prevention of angina pectoris due to coronary artery disease
Under development
Ondansetron
Prevent nausea and vomiting
Under development
Pentazocine
Analgesic drug
Under development
Illum (2002)
Propranolol hydrochloride
Management of hypertension and angina pectoris
Under development
Ranitidine hydrochloride
Treatment of duodenal ulcers
Under development
Behl et al. (1998)
Scopolamine
Motion sickness
Under development
Costantino et al. (2007)
Scopolamine hydrobromide
Prevention of nausea and vomiting induced
Under development
Behl et al. (1998)
Sufentanil
Analgesic drug
Under development
Behl et al. (1998)
Sumatriptan/Imitrex®
Migraine
Market
Grassin-Delyle et al. (2012)
Zanamivir
Antiviral drug
Under development
Zolmitriptan/Zomig®
Migraine
Market
Costantino et al. (2007)
Underlying this wide interest on exploiting nasal cavity for systemic delivery is the rapid and direct systemic absorption of compounds, the circumvention of gastrointestinal and hepatic first-pass metabolism and, consequently, the achievement of higher drug plasma levels and higher bioavailability through nasal route than oral administration. IN drug administration may enable the reduction of the dose administered, a quick onset of pharmacological activity and fewer side effects.
Among the several alternative formulations currently developed and under development, solution-based formulations are the most frequent because they are the easiest to administer and they have the greatest chance for systemic drug delivery across the nasal mucosa. Moreover, systems incorporating mucoadhesive excipients and/or enzyme inhibitors and/or nasal permeation enhancers have been developed in order to improve the therapeutic efficacy once they enhance drug nasal residence time, prolong duration of action and increase the absorption extent of drugs (Grassin-Delyle et al. 2012; Jiang et al. 2010; Pires et al. 2009).
15.3.1.1 Analgesic Drugs
Opioids are considered the cornerstone of an analgesic regimen and are indicated for the treatment of breakthrough pain and acute, moderate to severe and chronic pain. Ideally, they must exhibit a rapid onset time and a prolonged duration of action that coincides with the episode’s time course. Although oral and parenteral solutions are generally used for treatment of the breakthrough pain, the onset effect is not achieved before 30–45 min and the maximal effect within 1 h (Tveita et al. 2008). IN administration of opioids arises hence as a hope to easily and quickly achieve pain relief and improvement of the life quality of patients.
Indeed, a wide variety of opioid drugs have been under investigation, including morphine, fentanyl and buprenorphine. Although recognised as the standard opioid for cancer pain relief, morphine has a significant intestinal and hepatic first-pass metabolism that limits its bioavailability, which is around 20–32 % (Fitzgibbon et al. 2003). Similarly, the bioavailability of morphine solutions administered intranasally rounds only 10–22 % in humans and sheep (Illum et al. 2002) probably due to its low lipophilicity. In order to increase the nasal residence time, the bioavailability and the elimination half-life time of morphine after its IN administration, a wide variety of formulations have been currently under development, including formulations containing chitosan as microspheres or in solution (formulations based on starch microspheres coupled with lysophosphatidylcholine) (Illum et al. 2002) and solutions added of oleic acid as absorption promoter (Fitzgibbon et al. 2003). One of the most relevant clinical studies consisted in assessing the pharmacokinetic profile and tolerability of Rylominetm composed by morphine mesylate and chitosan in 13 subjects (Stoker et al. 2008). Based on the area under the concentration–time curve (AUC) values, bioavailability of IN morphine was considerably higher when compared to the other administration routes.
In opposition to morphine, fentanyl and butorphanol can be effectively and quickly absorbed at nasal cavity without using absorption promoters due to their relative high lipophilicity and low molecular weight. Particularly, IN fentanyl is currently marketed (Table 15.1) as two distinct forms: the aqueous solution Instanyl® and the pectin-based mucoadhesive formulation PecFent®. In a pharmacokinetic study in 19 cancer patients with breakthrough pain, nasal spray fentanyl was quickly absorbed through the nasal mucosa, attaining peak plasma concentrations within 12–15 min when administered at 50, 100 and 200 μg (Kaasa et al. 2010). One of the most important in vivo studies within this framework consisted in a balanced, randomised, double-blind, two-way crossover study in which patients received the same fentanyl dose by IN and IV administration (Christrup et al. 2008). The time to onset of action of around 10 min and the onset and duration of analgesia were not significantly different between single doses of IN and IV fentanyl in these adults. Recent researches have also shown an improvement of the bioavailability of fentanyl when administered as IN mucoadhesive formulations (Fisher et al. 2010; Kaasa et al. 2010).
Sumatriptan and zolmitriptan are analgesic drugs particularly used for migraine and cluster headaches. They are currently available as nasal formulations that provide onset times significantly quicker than those obtained after oral dosing (Dodick et al. 2005; Gawel et al. 2005) (Table 15.1). This success is due to the high lipophilicity of sumatriptan and zolmitriptan that facilitate their systemic absorption through nasal respiratory mucosa (Uemura et al. 2005) but also due to their direct access to CNS as it is referred in Sect. 15.4.1.5.
15.3.1.2 Cardiovascular Drugs
For a long time, nasal administration has been investigated as an attractive route for administration of cardiovascular drugs such as propranolol, nifedipine, nitroglycerin and carvedilol (Costantino et al. 2007).
The IN dosing of propranolol provides a pharmacokinetic profile that is very similar to that of IV administration, specifically when regarding the onset time and bioavailability (Ahn et al. 1995).
Bioadhesive sodium alginate microspheres of metoprolol tartrate for IN systemic delivery were also investigated as an alternative therapy for the treatment of hypertension and angina pectoris (Rajinikanth et al. 2003). Promising results were found in rabbits and rats, with maximum plasma drug concentrations (C max) clearly higher after IN administration than those after oral administration.
Nifedipine is a calcium channel blocking agent frequently used for the treatment of angina pectoris and hypertension. Kubota et al. (2001) performed a crossover clinical study in order to investigate the optimal administration method of nifedipine for rapid management of hypertension. It is interesting to highlight that although the value of C max was clearly lower after IN administration of nifedipine than that obtained for oral administration, the mean serum concentration of nifedipine 5 min after IN administration was higher (and remained higher until after 15 min). These results sustain that IN administration of nifedipine guarantees the fastest increase of drug plasma concentrations and the most significant effect on blood pressure reduction.
More recently, IN administration of carvedilol, a non-selective β-adrenergic antagonist also used in the treatment of hypertension and stable angina pectoris (Packer et al. 2002), has been under investigation due to its significant hepatic first-pass metabolism and low absolute bioavailability (25 %). Recent investigations reported that when administered by IN route to rabbits, sodium alginate microspheres and mucoadhesive chitosan microspheres containing carvedilol, the mean residence and half-life times of the drug were at least twice of those observed after IV administration. Furthermore, the high absolute bioavailability and the low t max achieved for carvedilol sustain that both pharmaceutical formulations are promissory to prolong the therapeutic effect of carvedilol (Patil et al. 2010, 2012).
15.3.1.3 Antiviral Drugs
The antiviral acyclovir is currently available as several dosage forms that present limitations. Firstly, the intestinal absorption of acyclovir is slow, variable and incomplete, with an absolute bioavailability of approximately 15–20 % which requires a frequent oral dose regimen. On the other hand, its low solubility in water and lipids hamper the administration of acyclovir by intramuscular route (Shao et al. 1994). Even when intravenously administered, acyclovir is mainly excreted unchanged through urine by glomerular filtration and tubular secretion, demanding a high dose to be administered in order to attain therapeutic drug concentrations.
Hence, the IN administration of acyclovir emerged recently as an innovative strategy that could maintain the drug for a longer time in systemic circulation within effective and non-toxic concentration ranges (Alsarra et al. 2008). Since acyclovir is also practically impermeable through the nasal mucosa, neutral mucoadhesive liposomes were formulated in order to enhance the nasal penetration and systemic absorption. In a study performed in rabbits, the absolute bioavailability of nasal liposomes with acyclovir was 60.7 % while that of free acyclovir was only around 5 %. This discrepancy was also observed for AUC values, clearly demonstrating that liposomes pass directly into systemic circulation, resulting in a considerable systemic concentration of acyclovir (Alsarra et al. 2008).
Similarly, zanamivir is another antiviral drug which, although presenting higher bioavailability when administered by IN route than orally (Cass et al. 1999), is poorly absorbed at nasal level especially due to its high hydrophilicity. Thus, similar investigations to those executed for acyclovir are expected to be soon performed for zanamivir.
15.3.1.4 Antiemetic and Motion Sickness Drugs
The nasal delivery of drugs for the treatment of nausea and motion sickness is steadily appearing as a desirable alternative to parenteral and oral medications especially because a rapid onset of action is required in acute situations. Moreover, the gastric dysmotility associated to the pathological situation is probable to affect the intestinal drug absorption and the drug fraction that is absorbed after oral administration.
For instance, when orally administered, metoclopramide bioavailability is highly variable (32–98 %) and it has a short half-life (3–4 h) that demands an oral administration three to four times daily. The IN administration of metoclopramide is identified as a good alternative (Mahajan and Gattani 2010). There are, however, limitations related to the low permeability across the nasal mucosa and the rapid MCC of metoclopramide, and in order to overcome these features, new nasal formulations have been developed and are under investigation. They consist on aqueous solutions added of absorption enhancers to increase nasal permeability (Zaki et al. 2006) or on gel and mucoadhesive formulations to prolong the residence time at the nasal absorption local and facilitate the drug uptake (Mahajan and Gattani 2010; Tas et al. 2009). Zaki et al. (2006) demonstrated that when nasal spray solution was administered to humans, the C max of metoclopramide was significantly higher than that observed after oral administration while values of t max and half-life time were significantly lower (Zaki et al. 2006). However, no statistical differences were observed for the mean residence times of metoclopramide, and therefore, the same research group developed and administered gel and mucoadhesive formulations composed by gellan gum (0.4 %, w/v) and Carbopol (0.15 %, w/v) to rabbits. The superior absolute bioavailability of the nasal gel compared to the oral solution clearly indicated higher absorption of metoclopramide when administered intranasally. Favourable results were also found for gel dosage forms based on mucoadhesive polymer sodium carboxymethylcellulose for IN administration of metoclopramide to sheep (Tas et al. 2009).
Ondansetron has also been under investigation to be administered by IN route, although it is currently available in IV solutions and oral dosage forms. The low oral bioavailability of ondansetron in humans (60 %) and its administration at least 30 min prior to chemotherapy sessions (Gungor et al. 2010) propelled Hussain and collaborators (2000) to investigate for the first time the feasibility of ondansetron IN administration to rats. The plasma concentration-time profiles for IN administration were comparable to that of IV administration and the rapid absorption through the nasal mucosa allowed ondansetron to reach systemic circulation almost instantaneously. Equivalent results were also reported by Gungor et al. (2010). Nevertheless, several ondansetron formulations have been developed and demonstrated to enhance drug delivery, reduce the onset time and prolong drug effect duration in relation to the oral administration (Cho et al. 2008; Gungor et al. 2010)
Scopolamine, an antimuscarinic agent indicated for motion sickness, is another example of a drug in this area that is suitable for IN dosing as depicted by human pharmacokinetic studies developed by Ahmed et al. 2000.
15.3.1.5 Erectile Dysfunction Drugs
Sildenafil citrate is considered a standard treatment for erectile dysfunction. It is rapidly absorbed after oral administration but only with an absolute bioavailability of 40 %, an onset of action time within 15.5 min and effect duration of approximately 40 min (Deveci et al. 2004). Recently, Elshafeey et al. (2009) attempted to take advantage of nasal administration to improve these limitations and developed a new microemulsion of sildenafil citrate composed of oleic acid/Labrasol/Transcutol/water. The research group achieved drug concentrations that were nearly twofold higher than those obtained for oral tablets. A higher bioavailability and faster onset systemic levels were also observed for IN formulation probably due to the fact that liver metabolism was bypassed.
15.4 Intranasal Delivery of CNS-Acting Drugs
The brain is a delicate organ that plays a set of vital functions to maintain convenient body homeostasis; therefore, its integrity is ensured by specific physiological barriers and mechanisms of defence which efficiently protect and isolate the CNS from harmful endogenous substances and external insults (e.g. xenobiotics and virus).
The BBB represents one of the strictest structural and functional barriers in segregating the brain from the systemic circulation. It is characterised by the presence of non-fenestrated capillary endothelial cells with intercellular tight junctions, a very high transendothelial electric resistance (Misra et al. 2003; Vyas et al. 2005a) and a high metabolic activity associated to the expression of numerous carrier-mediated efflux transporters (Anderson 1996; Rautio et al. 2008) that regulate the influx and efflux of a variety of compounds. Unfortunately, the CNS delivery of proficuous therapeutic agents is also frequently prevented. In the last decades, several different approaches have been attempted in order to circumvent the BBB and to deliver drugs efficiently to the brain for therapeutic or diagnostic applications (Illum 2000). For example, recent developments have generated much interest in the possibility of exploiting the IN administration as a non-invasive alternative route for delivery of drugs to the CNS. In fact, assuming the olfactory region as a unique direct connection between the nose and the brain, the IN administration has emerged as a promising approach for the delivery of therapeutic agents to the CNS bypassing the BBB (Hanson and Frey 2008; Illum 2004; Vyas et al. 2005a).
In many CNS disorders, a rapid and/or specific targeting of drugs to the brain would be beneficial. Therefore, valuable efforts have been conducted to improve brain delivery of various therapeutic agents via the IN route, in order to provide higher drug bioavailability at the biophase and consequently better therapeutic efficacy.
15.4.1 Nose-to-Brain Drug Delivery
IN drug administration provides a promising method to deliver therapeutics from the nasal cavity directly to the CNS, bypassing the BBB. Indeed, IN delivery represents an attractive alternative to oral and parenteral routes since, in addition to being non-invasive, it also allows the avoidance of gastrointestinal destruction and hepatic first-pass metabolism. Direct transport of drugs to the brain may lead to reducing systemic exposure and peripheral side effects, which allows the decrease of the dose and frequency of dosing as well as minimises toxicity and improves therapeutic efficacy by achieving desired drug concentrations at the biophase (Kumar et al. 2008; Seju et al. 2011). In addition, the rapid onset delivery of drugs to the CNS and the higher brain uptake congregate the essential conditions for the application of the IN route in the management of emergency situations (Florence et al. 2011; Li et al. 2002; Vyas et al. 2006a; Wolfe and Bernstone 2004).
The possible transport pathways by which a drug can be delivered to the CNS after IN administration are schematically depicted in Fig. 15.1. In general, therapeutic agents can travel from the nasal cavity to the brain via the olfactory route by two possible mechanisms: the olfactory epithelial pathway and the olfactory neural pathway (Merkus and Van den Berg 2007). Similar to drug absorption through nasal respiratory mucosa, in the olfactory epithelial pathway, drugs can be absorbed across the olfactory epithelium either by transcellular or paracellular transport.
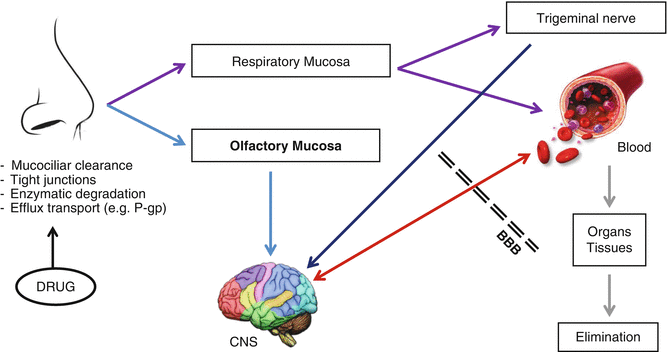
Fig. 15.1
Schematic representation of the possible pathways involved in the transport of drugs from nose to brain
In the olfactory neural pathway, drugs can be transferred via axonal internalisation with subsequent transport along the olfactory sensory nerves directly to the brain. Nevertheless, it is believed that such transport is slow, taking hours or even days for drugs to reach the brain parenchymal tissue (Dhuria et al. 2010; Thorne and Frey 2001). As an alternative, it was suggested that drugs after traversing the olfactory epithelium could make their way by paracellularly entering into the perineuronal channels that surround the olfactory nerves, requiring only few minutes (<30 min) to travel along the olfactory axon up to the cerebral spinal fluid (CSF) (Dhuria et al. 2010). Recently, trigeminal nerve pathway has also been advocated as another and additional valid route for the transport of molecules directly from the nasal cavity to the brain (Dhuria et al. 2010; Ross et al. 2004; Thorne et al. 2004).
The hypothetic mechanisms of direct delivery of drugs from nasal passages to the CNS were described; notwithstanding, the contributions underlying each one are not yet clearly elucidated. Generally, the rapid appearance of a drug in the brain and CSF indicates preferential involvement of extracellular transport pathways rather than the olfactory neural route. However, the possibility of occurring later axonal drug internalisation cannot be entirely ruled out. Nasally applied drugs could reach the CNS by means of one or a combination of various transport pathways (Fig. 15.1).
15.4.1.1 Alzheimer’s Disease Drugs
Several oral acetylcholinesterase inhibitors including rivastigmine, donepezil, galantamine and tacrine have been used for the treatment of Alzheimer’s disease symptoms. Notwithstanding, oral administration of such molecules has often been associated with low bioavailability, extensive first-pass metabolism, short elimination half-life, hepatotoxicity and severe gastrointestinal side effects (Costantino et al. 2008).
The potential of the IN delivery route for targeting acetylcholinesterase inhibitors to the brain seems to provide valuable benefits and has been investigated in animal models. The uptake of NXX-066 (a physostigmine analogue) in the CSF after nasal and IV administration to rats was investigated in order to assess whether a direct nose-to-brain pathway is involved (Dahlin and Björk 2001). Study results demonstrated that only low concentrations of NXX-066 were detected in the CSF following both routes of administration. However, nasal administration resulted in extremely rapid and complete absorption of NXX-066 into the systemic circulation exhibiting an absolute bioavailability near to 100 %. The high values of nasal bioavailability suggest that this route could be a suitable alternative to oral and parenteral administrations.
The concentrations of tacrine in blood and brain after IN and IV administration to mice were also evaluated by Jogani et al. (2007). Pharmacokinetic data revealed that drug concentrations in brain tissue were found to be significantly higher for IN administration and the delivery of nasal tacrine to the brain showed to be much quicker than given via the IV route. These findings demonstrated that after IN delivery, a preferential nose-to-brain transport is implied in the selective distribution of tacrine to the brain.
15.4.1.2 Parkinson’s Disease Drugs
Until now, there is no cure for Parkinson’s disease but its symptoms can be attenuated by the replacement of the dopamine basal levels at the brain. However, dopamine is unable to cross the BBB in appreciable amounts making its administration via oral and parenteral routes not feasible. Therefore, levodopa (L-dopa) is currently the gold standard treatment in Parkinson’s disease, since it easily penetrates the BBB and is rapidly converted to dopamine within the brain. Unfortunately, the clinical response to oral L-dopa is commonly variable and unreliable, due to its erratic absorption and first-pass metabolism (Kao et al. 2000). Additionally, about 95 % of the drug undergoes decarboxylation to dopamine in the peripheral tissues (Dahlin et al. 2000), compromising the amount of unchanged drug available to reach the brain and enhancing the occurrence of adverse effects. In this context, the transfer of dopamine along the olfactory pathway to the CNS following nasal administration has been assessed in rodents (Dahlin et al. 2000, 2001). The experimental results showed that there was an effective transport of dopamine from the nasal cavity into the CNS, since concentration levels after nasal administration were, in comparison to IV injection, 2.3 and 6.8 times higher in the CSF and olfactory bulb, respectively (Dahlin et al. 2000). Nevertheless, the fraction of the nasally administered drug that reached the brain tissue was only 0.12 % of the total dose, suggesting that higher doses of dopamine may be required to guarantee therapeutic efficacy (Dahlin et al. 2000).
The potential of direct nose-to-brain transport of L-dopa was also investigated in rats. Although the AUC values of nasal L-dopa were more than two times higher in plasma and brain comparatively to oral administration, a large fraction of drug was systemically absorbed via the nasal route, and therefore, the fraction of drug transported by the direct nose-to-brain pathway was minimal (Kim et al. 2009). More promising results were achieved by Kao et al. (2000) using the prodrug approach. Following IN administration of the butyl ester prodrug of L-dopa, CNS bioavailability of L-dopa was improved comparing to an equivalent dose given intravenously.
15.4.1.3 Anticonvulsant and Antiepileptic Drugs
Oral administration of anticonvulsant drugs has generally been associated with high systemic distribution into nontargeted tissues, peripheral adverse effects and limited brain uptake. Moreover, patient’s physical condition immediately after a convulsive episode is incompatible with the oral ingestion of a tablet dosage form. Apart from its advantages on the clinical emergencies in acute seizure situations, nasally administered anticonvulsant drugs may represent a valuable approach for the long-term treatment of epilepsy by providing the decrease of the dose, frequency of dosing and related side effects thus improving therapeutic efficacy and tolerability.
IV benzodiazepines, such as diazepam, lorazepam, midazolam and clonazepam, have been used as the first-line therapy for the termination of seizure activity in status epilepticus. However, benzodiazepines IV dosing may unleash hypotension, cardiac dysrhythmia and respiratory failure (Li et al. 2000). Aiming to minimise the disadvantages and potentiate the therapeutic index of such drugs, several studies were carried out on the subject of IN delivery. A comparative study between IV injection and three nasal formulations of clobazam (solution, microemulsion and mucoadhesive microemulsion) was performed in mice in order to assess and characterise its pharmacokinetic profile and pharmacodynamic performance (Florence et al. 2011). The pharmacokinetic results revealed that the systemic blood distribution of the drug was significantly lower with IN-administered formulations comparatively to IV injection, thus ensuring drug targeting at the site of action and minimising the possibility of systemic side effects. Furthermore, higher brain AUC and C max for microemulsion formulations reflect an enhanced CNS uptake, indicating that a preferential nose-to-brain transport may be involved, revealing consistency with similar previous studies with clonazepam (Vyas et al. 2006a). By virtue of their lipophilic nature and lower interfacial tension, microemulsions heighten the drug permeability across the nasal mucosa. On the other hand, the incorporation of a mucoadhesive agent (Carbopol) improves drug uptake by opening tight junctions, increasing paracellular transport of the molecules.
To investigate brain targeting via nasal administration, the antiepileptic drug carbamazepine (CBZ) was chosen as a model. Taking into account that CBZ is absorbed slowly and erratically after oral administration, displays a bioavailability of less than 50 % and usually attains peak plasma concentration 4–8 h after oral ingestion (Barakat et al. 2006), a direct delivery of this drug to the brain circumventing the BBB would be highly beneficial. In this context, a CBZ gel formulation composed by hypromellose and Carbopol 974P (3:1) was nasally administered to rats, aiming to compare CBZ concentrations in blood and brain tissue samples with other conventional routes, such as oral and IV administration (Barakat et al. 2006). Experimental data revealed that IN CBZ concentrations were greater in brain than in plasma, also achieving remarkably higher levels in CNS compared to oral or IV administration. A direct transport pathway from nose to brain was demonstrated since peak brain concentration after nasal administration was attained in only 5 min and CBZ absorption from the nasal cavity into the brain was rapid and complete.
15.4.1.4 Analgesic Drugs
Nasal administration of morphine is currently under development in order to overcome its extensive hepatic first-pass effect, affording a more rapid drug absorption and faster onset of action. Indeed, systemic absorption of morphine after nasal administration undoubtedly contributes to achieve these goals as already stated in Sect. 15.3.1.1 . However, taking into account that morphine is a small hydrophilic molecule with limited BBB permeability, direct transport of the drug along the olfactory pathway from nose to the brain would be advantageous for pain relief. For these reasons, some investigations have been carried out in order to evaluate the direct access of morphine to the brain. Following IN administration of morphine to rodents, Westin and collaborators (2005) found that the drug was rapidly transferred via the olfactory epithelium to the CNS, reaching the highest concentration in the olfactory bulb after 15 and 60 min in rats and mice, respectively. Upon these facts, the same research group intended to quantify the olfactory transfer of morphine to the brain by comparing drug levels in brain and plasma after both IN and IV administration (Westin et al. 2006). The results showed that after nasal and IV administration of the same dose (1 mg/kg body weight), equal morphine concentrations were obtained in the brain at 5 and 15 min. However, brain to plasma AUC ratio from 0 to 5 min was substantially higher for nasal delivery compared to IV infusion, proving an early distribution of morphine to the CNS via the nasal route.
15.4.1.5 Migraine and Cluster Headaches Drugs
Sumatriptan and zolmitriptan are the drugs most commonly used in the effective treatment of migraine and cluster headaches (Jain et al. 2010). Although these drugs present potent analgesic activity on acute migraine pain relief, current oral therapies are commonly associated with a slow onset of action and significant hepatic first-pass metabolism which results in low absolute plasma bioavailability (Jain et al. 2010; Vyas et al. 2005b). Furthermore, the majority of migraine patients experience several gastrointestinal disturbances during the attacks making the intake of oral tablets often inappropriate (Yates et al. 2005).
Although systemic absorption of IN sumatriptan and zolmitriptan is undeniable, their eventual transport from the nasal cavity directly to the brain may also have an important contribution for the treatment of migraine and cluster headaches. Therefore, the assessment of nose-to-brain delivery of IN mucoadhesive microemulsions of both sumatriptan and zolmitriptan has been investigated in rats (Vyas et al. 2005b, 2006b). The mucoadhesive microemulsions showed better results than microemulsions or drug solutions given nasally. Superior pharmacokinetic results were even attained for the developed sumatriptan microemulsions compared to an already marketed nasal product (Vyas et al. 2006b). Comparatively to IV administration, higher C max and AUC values were found in the brain at all sampling time points for nasally administered formulations, suggesting that preferential nose-to-brain transport may be attributed to both drugs. These findings were also sustained by Jain et al. (2010) who demonstrated that zolmitriptan is predominantly transported to the brain via the olfactory and trigeminal pathways.
15.4.1.6 Antipsychotic and Antidepressant Drugs
Atypical antipsychotic drugs are currently the first choice for the treatment of schizophrenia, and they are available in the market predominantly under oral dosage forms. Oral formulations are, however, related to low plasma drug bioavailability which frequently demands the increase of dose and frequency of dosing. As a consequence, the occurrence of adverse effects is also often potentiated. In this context, the development of IN delivery systems of several antipsychotic agents like risperidone and olanzapine has been attempted considering the potential of this route for direct brain targeting (Kumar et al. 2008; Seju et al. 2011). Promising results were obtained for the IN delivery experiments of a mucoadhesive nanoemulsion of risperidone- and olanzapine-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles using animal models. Higher drug concentrations were observed in the brain for the developed formulations compared to the plain solution of the drug given either nasally or intravenously. Pharmacokinetic data of olanzapine-loaded PLGA nanoparticles even showed an additional therapeutic gain by providing sustained drug delivery to the brain (Seju et al. 2011). In fact, the nanoparticle strategy offers an improvement in nose-to-brain delivery since, in addition to protecting the encapsulated drug from biological or chemical degradation and efflux P-glycoprotein (P-gp) transport, it also enables the increase of the drug residence time within the nasal cavity. As a result, the opportunity to provide sustained delivery of olanzapine is increased, allowing the enhancement of brain drug concentrations.
An IN delivery system of milnacipran was also investigated for the treatment of depression (Uchida et al. 2011). A pharmacokinetic assessment of plasma and CSF milnacipran concentrations following nasal drug delivery to rats revealed that, in comparison to intraduodenal administration, higher C max and lower t max were observed for both matrices. These pharmacokinetic data were in agreement with the results obtained for the pharmacodynamic evaluations in which the antidepressant effect after IN administration of milnacipran was higher and quicker than after oral dosing. The impact of the co-administration of IN milnacipran with 0.5 % chitosan was also addressed in this study. The incorporation of this polysaccharide into the nasal formulation led to an even greater antidepressant effect since it provided a long residence time of milnacipran within the nasal cavity, thus resulting in the increase of the systemic absorption as well as direct transport of the drug to the CNS.
15.4.1.7 Antiviral Drugs
The efficacy of antiviral therapy in the treatment of neuroinfections is often limited due to reduced drug uptake into the CNS as a consequence of its poor permeation across the BBB (Colombo et al. 2011). Indeed, most of the antiviral agents are highly hydrophilic compounds and therefore cannot passively diffuse through the BBB easily. Moreover, it is estimated that a huge part of them are also substrates of the P-gp efflux pump (Hanson and Frey 2007) which has a markedly role on CNS protection by hindering the access of a wide variety of substances to the brain.
Several studies have recently investigated the pharmacokinetics and brain distribution profiles of some antiviral agents after nasal and IV administration to animal models. A preferential transfer of zidovudine, a reverse transcriptase inhibitor, into the CSF and brain tissues following IN administration to rabbits was successfully demonstrated, providing a promising therapeutic option for the treatment of CNS dysfunctions caused by human immunodeficiency virus (HIV) (Ved and Kim 2011). By using a thermo-reversible gelling system comprising Poloxamer 407 as a mucoadhesive polymer and n-tridecyl-β-D-maltoside as a permeation enhancer, the authors guaranteed a larger increase of zidovudine brain bioavailability relatively to solutions given both nasally and intravenously. The existence of a direct nose-to-brain pathway to transport zidovudine from the nasal cavity to the CNS was also strongly proven. According to Ved and Kim (2011), approximately 99 % of zidovudine content was directly transferred to the brain via the olfactory route.
15.5 Intranasal Delivery of Biomacromolecular Drugs
IN administration represents a promising choice for delivery of a variety of high molecular weight therapeutic agents such as peptide-, protein- or nucleic acid-based drugs (Csaba et al. 2009; Singh et al. 2012). Because of the higher susceptibility of biological therapies to enzymatic degradation and due to their low permeability across the epithelium via transcellular and paracellular pathways, the absorption of these biomacromolecular drugs from mucosal sites is poor. Therefore, to increase their bioavailability, they are mostly administered by parenteral routes. Over the last years, new pharmaceutical formulations and novel delivery strategies have been developed offering promising opportunities to expand the IN delivery of biomacromolecules (Ozsoy et al. 2009; Singh et al. 2012).
As the nasal mucosa is one of the most permeable and highly vascularised tissues, also avoiding gastrointestinal and hepatic first-pass metabolism, the extent of absorption of biomacromolecules may be potentiated by IN administration comparatively to that achieved through oral route. Accordingly, the nasal route has gained a great interest as an alternative and non-invasive way for systemic and/or direct brain delivery of various classes of biological therapeutic agents (Ozsoy et al. 2009; Veronesi et al. 2011).
The recent advances in the field of biotechnology have promoted the emergence of a range of biodrugs. Besides therapeutic peptides and proteins, a broad variety of other biodrugs are coming into clinical practice or moving to a greater extent into clinical research, namely, vaccines, cell or gene therapies, cytokines, tissue growth factors and monoclonal antibodies (Csaba et al. 2009; Ozsoy et al. 2009; Singh et al. 2012). Hence, it is expected that the number of biomacromolecular drugs commercially available for administration via nasal route will progressively increase.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


