Neuro-Ophthalmology
10.1 ANISOCORIA
Eyelid position and extraocular motility MUST be evaluated when anisocoria is present (see Figure 10.1.1).
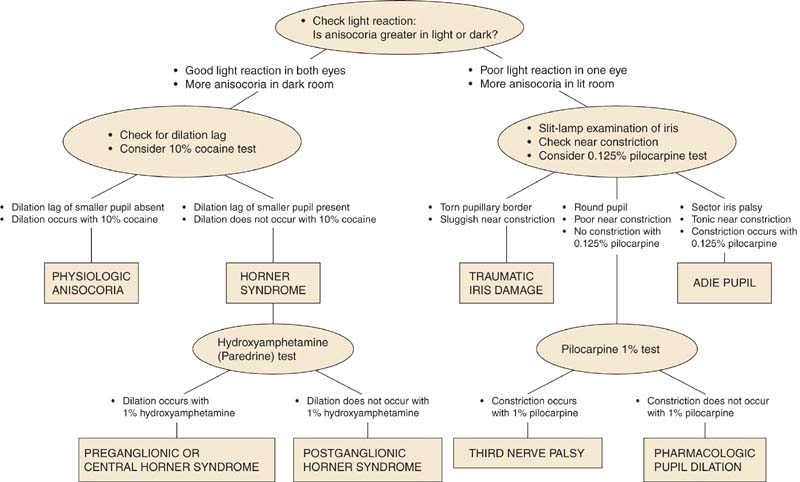
FIGURE 10.1.1. Flow diagram for the work-up of anisocoria. (Modified from Thompson HS, Pilley SF. Unequal pupils. A flow chart for sorting out the anisocorias. Surv Ophthalmol 1976;21:45–48, with permission.)
Classification
1. The abnormal pupil is constricted.
—Unilateral exposure to a miotic agent (e.g., pilocarpine).
—Iritis: Eye pain, redness, and anterior chamber cells and flare.
—Horner syndrome: Mild ptosis on the side of the small pupil. See 10.2, Horner Syndrome.
—Argyll Robertson (syphilitic) pupil: Always bilateral, irregularly round miotic pupils, but a mild degree of anisocoria is often present. See 10.3, Argyll Robertson Pupils.
—Long-standing Adie pupil: The pupil is initially dilated, but over time may constrict. Hypersensitive to pilocarpine 0.125%. See 10.4, Adie (Tonic) Pupil.
2. The abnormal pupil is dilated.
—Iris sphincter muscle damage from trauma: Torn pupillary margin or iris transillumination defects seen on slit-lamp examination.
—Adie (tonic) pupil: The pupil may be irregular, reacts minimally to light, and slowly and tonically to convergence. Hypersensitive to pilocarpine 0.125%. See 10.4, Adie (Tonic) Pupil.
—Third cranial nerve palsy: Always associated ptosis and/or extraocular muscle palsies. See 10.5, Isolated Third Nerve Palsy.
—Unilateral exposure to a mydriatic agent: Cycloplegic drops (e.g., atropine), scopolamine patch for motion sickness, ill-
fitting mask in patients on nebulizers (using ipratropium bromide), possible use of sympathetic medications (e.g., pseudoephedrine). If the mydriatic exposure is recent, pupil will not react to pilocarpine 1%.
3. Physiologic anisocoria: Pupil size disparity is the same in light as in dark, and the pupils react normally to light. The size difference is usually, but not always, <2 mm in diameter.
Work-Up
1. History: When was the anisocoria first noted? Associated symptoms or signs? Ocular trauma? Eye drops or ointments? Syphilis? Old photographs?
2. Ocular examination: Try to determine which pupil is abnormal by comparing pupil sizes in light and in dark. Anisocoria greater in light suggests the abnormal pupil is the larger pupil; anisocoria greater in dark suggests the abnormal pupil is the smaller pupil. Test the pupillary reaction to both light and near. Evaluate for the presence of a relative afferent pupillary defect. Look for ptosis, evaluate ocular motility, and examine the pupillary margin with a slit lamp.
—If the abnormal pupil is small, a diagnosis of Horner syndrome may be confirmed by a cocaine or apraclonidine test (see 10.2, Horner Syndrome). In the presence of ptosis and an unequivocal increase in anisocoria in dim illumination, cocaine and apraclonidine testing may be unnecessary.
—If the abnormal pupil is large and there is no sphincter muscle damage or signs of third nerve palsy (e.g., extraocular motility deficit, ptosis), the pupils are tested with one drop of pilocarpine 0.125%. Within 10 to 15 minutes, an Adie pupil will constrict significantly more than the fellow pupil [see 10.4, Adie (Tonic) Pupil].
NOTE: For an acute Adie pupil, the pupil may not react to a weak cholinergic agent.
—If the pupil does not constrict with pilocarpine 0.125%, or pharmacologic dilatation is suspected, pilocarpine 1% is instilled in both eyes. A normal pupil constricts sooner and to a greater extent than the pharmacologically dilated pupil. An eye that recently received a strong mydriatic agent such as atropine usually will not constrict at all.
See 10.2, Horner Syndrome, 10.3, Argyll Robertson Pupils, 10.4, Adie (Tonic) Pupil, and 10.5, Isolated Third Nerve Palsy.
10.2 HORNER SYNDROME
Symptoms
Ptosis and anisocoria. May have anhydrosis. Often asymptomatic.
Signs
(See Figure 10.2.1.)

FIGURE 10.2.1. Right Horner syndrome with ptosis and miosis.
Critical. Anisocoria that is greater in dim illumination (especially during the first few seconds after the room light is dimmed). The abnormal small pupil dilates less than the normal, larger pupil. Mild ptosis (2 mm) and lower eyelid elevation (“reverse ptosis”) occur on the side of the small pupil.
Other. Lower intraocular pressure, lighter iris color in congenital cases (iris heterochromia), loss of sweating (anhydrosis, distribution depends on the site of lesion), transient increase in accommodation (older patients hold their reading card closer in the Horner eye). Involved eye may have conjunctival hyperemia due to decreased episcleral vascular tone. Light and near reactions are intact.
Differential Diagnosis
See 10.1, Anisocoria.
Etiology
 First-order neuron disorder: Stroke (e.g., vertebrobasilar artery insufficiency or infarct), tumor, multiple sclerosis (MS). Rarely, severe osteoarthritis of the neck with bony spurs.
First-order neuron disorder: Stroke (e.g., vertebrobasilar artery insufficiency or infarct), tumor, multiple sclerosis (MS). Rarely, severe osteoarthritis of the neck with bony spurs.
 Second-order neuron disorder: Tumor (e.g., lung carcinoma, metastasis, thyroid adenoma, neurofibroma). Patients with pain in the arm or scapular region should be suspected of having a Pancoast tumor. In children, consider neuroblastoma, lymphoma, or metastasis.
Second-order neuron disorder: Tumor (e.g., lung carcinoma, metastasis, thyroid adenoma, neurofibroma). Patients with pain in the arm or scapular region should be suspected of having a Pancoast tumor. In children, consider neuroblastoma, lymphoma, or metastasis.
 Third-order neuron disorder: Headache syndrome (e.g., cluster, migraine, Raeder paratrigeminal syndrome), internal carotid dissection, varicella zoster virus, otitis media, Tolosa–Hunt syndrome, neck trauma/tumor/inflammation, cavernous sinus pathology.
Third-order neuron disorder: Headache syndrome (e.g., cluster, migraine, Raeder paratrigeminal syndrome), internal carotid dissection, varicella zoster virus, otitis media, Tolosa–Hunt syndrome, neck trauma/tumor/inflammation, cavernous sinus pathology.
 Congenital Horner syndrome: May also be caused by birth trauma.
Congenital Horner syndrome: May also be caused by birth trauma.
 Other rare causes: Cervical paraganglioma, ectopic cervical thymus.
Other rare causes: Cervical paraganglioma, ectopic cervical thymus.
Work-Up
1. Diagnosis confirmed with a positive cocaine test. Place one drop of 10% cocaine in both eyes. Check in 15 minutes. If no change in pupillary size is noted, repeat drops and recheck the pupils in 15 minutes (repeat until normal pupil dilates). A Horner pupil dilates less than the normal pupil. Alternatively, either 1% or 0.5% apraclonidine may be used. Apraclonidine causes relative reversal of anisocoria. The miotic pupil with Horner syndrome will appear larger than the normal pupil.
NOTE: There may be a high false-negative rate to pharmacologic testing in an acute Horner syndrome.
2. A third-order neuron disorder may be distinguished from a first- and second-order neuron disorder with hydroxyamphetamine. Place one drop of 1% hydroxyamphetamine in both eyes. Check in 15 minutes and repeat if no change in pupillary size is noted. Failure of the Horner pupil to dilate to an equivalent degree as the fellow eye indicates a third-order neuron lesion, which may help guide the work-up. However, most experts feel the entire sympathetic pathway should be imaged in Horner syndrome regardless of the results of pharmacologic testing.
NOTE: The hydroxyamphetamine test has a sensitivity of up to 93% and a specificity of 83% for identifying a third-order neuron lesion. Hydroxyamphetamine should not be used within 24 hours of cocaine or apraclonidine to avoid possible interference with each other. Both drops require an intact corneal epithelium and preferably no prior eye drops (including anesthetic drops) for accurate results.
3. Determine the duration of the Horner syndrome from the patient’s history and an examination of old photographs. New-onset Horner syndrome requires a more urgent diagnostic work-up to exclude life-threatening etiologies [(e.g., internal carotid artery dissection, which can present with transient visual loss, head/neck/face pain, or dysgeusia (foul taste in the mouth)]. An old Horner syndrome is more likely to be benign.
4. History: Headaches? Arm pain? Previous stroke? Previous surgery that may have damaged the sympathetic chain, including cardiac, thoracic, thyroid, or neck surgery? History of head or neck trauma? Ipsilateral neck pain?
5. Physical examination (especially check for supraclavicular nodes, thyroid enlargement, or a neck mass).
6. Complete blood count (CBC) with differential.
7. Computed tomography (CT) of the chest to evaluate lung apex for possible mass (e.g., Pancoast tumor).
8. Magnetic resonance imaging (MRI) of the brain and neck.
9. Magnetic resonance angiography (MRA) or CT angiography (CTA) of head/neck or carotid Doppler ultrasonography (US) to evaluate for carotid artery dissection (especially with neck pain). MRA and CTA appear to be superior to Doppler US for evaluation of carotid artery dissection and allow for evaluation of the distal internal carotid artery. Additionally, imaging of the brain can be accomplished during the same examination which streamlines the work-up process. Obtain carotid angiography if MRA, CTA, or Doppler yield equivocal results.
10. Lymph node biopsy when lymphadenopathy is present.
Treatment
1. Treat the underlying disorder if possible.
NOTE: Carotid dissection usually requires anticoagulation or antiplatelet therapy to prevent carotid occlusion and hemispheric stroke in consultation with Neurology and Neurosurgery. Rarely, ischemic symptoms in the distribution of the dissection persist despite anticoagulation. In these cases, surgical intervention may be considered.
2. Ptosis surgery may be performed electively.
Follow-Up
1. Work-up acute Horner syndromes as soon as possible to rule out life-threatening causes. MRA should be performed the same day for dissection (it is convenient to perform an MRI simultaneously). The other tests should be performed within 1 to 2 days.
2. Chronic Horner syndrome can be evaluated with less urgency. There are no ocular complications that necessitate close follow-up.
10.3 ARGYLL ROBERTSON PUPILS
Symptoms
Usually asymptomatic.
Signs
Critical. Small, irregular pupils that exhibit “light-near” dissociation (react poorly or not at all to light but constrict normally during convergence). By definition, vision must be intact.
Other. The pupils dilate poorly in darkness. Always bilateral, although may be asymmetric.
Differential Diagnosis of “Light-Near” Dissociation
 Bilateral optic neuropathy or severe retinopathy: Reduced visual acuity with normal pupil size.
Bilateral optic neuropathy or severe retinopathy: Reduced visual acuity with normal pupil size.
 Adie (tonic) pupil: Unilateral or bilateral irregularly dilated pupil that constricts slowly and unevenly to light. Normal vision. See 10.4, Adie (Tonic) Pupil.
Adie (tonic) pupil: Unilateral or bilateral irregularly dilated pupil that constricts slowly and unevenly to light. Normal vision. See 10.4, Adie (Tonic) Pupil.
 Dorsal midbrain (Parinaud) syndrome: Bilateral, normal-to-large pupils. May be accompanied by convergence retraction nystagmus, lid retraction, and supranuclear upgaze palsy. See 10.4, Adie (Tonic) Pupil and “Convergence-retraction” in 10.21, Nystagmus.
Dorsal midbrain (Parinaud) syndrome: Bilateral, normal-to-large pupils. May be accompanied by convergence retraction nystagmus, lid retraction, and supranuclear upgaze palsy. See 10.4, Adie (Tonic) Pupil and “Convergence-retraction” in 10.21, Nystagmus.
 Rarely caused by third nerve palsy with aberrant regeneration. See 10.6, Aberrant Regeneration of the Third Nerve.
Rarely caused by third nerve palsy with aberrant regeneration. See 10.6, Aberrant Regeneration of the Third Nerve.
 Others: Diabetes, alcoholism, etc.
Others: Diabetes, alcoholism, etc.
Etiology
 Tertiary syphilis.
Tertiary syphilis.
Work-Up
1. Test the pupillary reaction to light and convergence: To test the reaction to convergence, patients are asked to look first at a distant target and then at their own finger, which the examiner holds in front of them and slowly brings in toward their face.
2. Slit-lamp examination: Look for interstitial keratitis (see 4.18, Interstitial Keratitis).
3. Dilated fundus examination: Search for chorioretinitis, papillitis, and uveitis.
4. Fluorescent treponemal antibody absorption (FTA-ABS) or microhemagglutination-Treponema pallidum (MHA-TP), rapid plasma reagin (RPR) or Venereal Disease Research Laboratories (VDRL) test.
5. If the diagnosis of syphilis is established, lumbar puncture (LP) may be indicated. See 12.12, Syphilis, for specific indications.
Treatment
1. Treatment based on the presence of active disease and previous appropriate treatment.
2. See 12.12, Syphilis, for treatment indications and specific antibiotic therapy.
Follow-Up
This is not an emergency. Diagnostic work-up and determination of syphilitic activity should be undertaken within a few days.
10.4 ADIE (TONIC) PUPIL
Symptoms
Difference in size of pupils, blurred near vision, photophobia. May be asymptomatic.
Signs
Critical. An irregularly dilated pupil that has minimal or no reactivity to light. Slow, tonic constriction with convergence, and slow redilation.
NOTE: It is typically unilateral initially and more common in young women.
Other. May have an acute onset and become bilateral. The involved pupil may become smaller than the normal pupil over time.
Differential Diagnosis
See 10.1, Anisocoria.
 Parinaud syndrome/dorsal midbrain lesion: Bilateral mid-dilated pupils that react poorly to light but constrict normally with convergence (not tonic). Associated with eyelid retraction (Collier Sign), supranuclear upgaze paralysis, and convergence retraction nystagmus. An MRI should be performed to rule out pinealoma and other midbrain pathology.
Parinaud syndrome/dorsal midbrain lesion: Bilateral mid-dilated pupils that react poorly to light but constrict normally with convergence (not tonic). Associated with eyelid retraction (Collier Sign), supranuclear upgaze paralysis, and convergence retraction nystagmus. An MRI should be performed to rule out pinealoma and other midbrain pathology.
 Holmes–Adie syndrome: Tonic pupil and tendon areflexia. May be associated with autonomic and peripheral neuropathy.
Holmes–Adie syndrome: Tonic pupil and tendon areflexia. May be associated with autonomic and peripheral neuropathy.
Etiology
Idiopathic most commonly. Orbital trauma, surgery, and varicella zoster infection are seen frequently. Early syphilis, parvovirus B19, herpes simplex virus, botulism, paraneoplastic syndrome, giant cell arteritis (GCA), pan-retinal photocoagulation, and neurological Lyme disease less commonly. Rare associations reported with endometriosis, seminomas, and Sjogren syndrome.
Work-Up
See 10.1, Anisocoria, for a general work-up when the diagnosis is uncertain.
1. Evaluate pupils at slit lamp or with a muscle light for irregular and slow constriction.
2. Test for cholinergic hypersensitivity. Instill 0.125% pilocarpine in both eyes and recheck pupils in 10 to 15 minutes. An Adie pupil constricts while the normal pupil does not.
3. If bilateral simultaneous Adie pupils, consider further laboratory investigations including testing for the aforementioned etiologies. For unilateral involvement, no further laboratory investigations are necessary.
NOTE: The dilute pilocarpine test may occasionally be positive in familial dysautonomia. Hypersensitivity may not be present with an acute Adie pupil and may need to be retested a few weeks later.
4. If Adie pupil is present in a patient younger than 1 year, consult a pediatric neurologist to rule out familial dysautonomia (Riley–Day syndrome).
Treatment
Pilocarpine 0.125% b.i.d. to q.i.d. for cosmesis and to aid in accommodation, if desired.
Follow-Up
If the diagnosis is certain, follow-up is routine.
10.5 ISOLATED THIRD NERVE PALSY
Symptoms
Binocular diplopia and ptosis; with or without pain.
NOTE: Pain does not distinguish between microvascular infarction and compression.
Signs
(See Figures 10.5.1 through 10.5.4.)
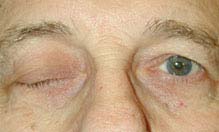
FIGURE 10.5.1. Isolated right third cranial nerve palsy with complete ptosis.
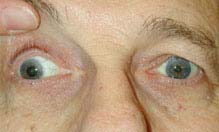
FIGURE 10.5.2. Isolated right third cranial nerve palsy: Primary gaze showing right exotropia and dilated pupil.
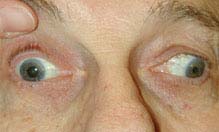
FIGURE 10.5.3. Isolated right third cranial nerve palsy: Left gaze showing inability to adduct right eye.
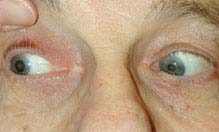
FIGURE 10.5.4. Isolated right third cranial nerve palsy: Right gaze showing normal abduction of right eye.
Critical.
1. External ophthalmoplegia.
—Complete palsy: Limitation of ocular movement in all fields of gaze except temporally.
—Incomplete palsy: Partial limitation of ocular movement.
—Superior division palsy: Ptosis and an inability to look up.
—Inferior division palsy: Inability to look nasally or inferiorly.
2. Internal ophthalmoplegia.
—Pupil-involving: A fixed, dilated, poorly reactive pupil.
—Pupil-sparing: Pupil not dilated and normally reactive to light.
—Relative pupil-sparing: Pupil partially dilated and sluggishly reactive to light.
Other. An exotropia or hypotropia. Aberrant regeneration. See 10.6, Aberrant Regeneration of the Third Nerve.
Differential Diagnosis
 Myasthenia gravis: Diurnal variation of symptoms and signs, pupil never involved, increased eyelid droop after sustained upgaze. See 10.11, Myasthenia Gravis.
Myasthenia gravis: Diurnal variation of symptoms and signs, pupil never involved, increased eyelid droop after sustained upgaze. See 10.11, Myasthenia Gravis.
 Thyroid eye disease: Eyelid lag, lid retraction, injection over the rectus muscles, proptosis, positive forced-duction testing. See 7.2.1, Thyroid Eye Disease.
Thyroid eye disease: Eyelid lag, lid retraction, injection over the rectus muscles, proptosis, positive forced-duction testing. See 7.2.1, Thyroid Eye Disease.
 Chronic progressive external ophthalmoplegia (CPEO): Bilateral, slowly progressive ptosis and motility limitation. Pupil spared, often no diplopia. See 10.12, Chronic Progressive External Ophthalmoplegia.
Chronic progressive external ophthalmoplegia (CPEO): Bilateral, slowly progressive ptosis and motility limitation. Pupil spared, often no diplopia. See 10.12, Chronic Progressive External Ophthalmoplegia.
 Idiopathic orbital inflammatory syndrome: Pain and proptosis common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
Idiopathic orbital inflammatory syndrome: Pain and proptosis common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
 Internuclear ophthalmoplegia (INO): Unilateral or bilateral adduction deficit with horizontal nystagmus of opposite abducting eye. No ptosis. See 10.13, Internuclear Ophthalmoplegia.
Internuclear ophthalmoplegia (INO): Unilateral or bilateral adduction deficit with horizontal nystagmus of opposite abducting eye. No ptosis. See 10.13, Internuclear Ophthalmoplegia.
 Skew deviation: Supranuclear brainstem lesion producing asymmetric, mainly vertical ocular deviation not consistent with single cranial nerve defect. See Differential Diagnosis in 10.7, Isolated Fourth Nerve Palsy.
Skew deviation: Supranuclear brainstem lesion producing asymmetric, mainly vertical ocular deviation not consistent with single cranial nerve defect. See Differential Diagnosis in 10.7, Isolated Fourth Nerve Palsy.
 Parinaud syndrome/dorsal midbrain lesion: Bilateral mid-dilated pupils that react poorly to light but constrict normally with convergence (not tonic). Associated with eyelid retraction (Collier Sign), supranuclear upgaze paralysis, and convergence retraction nystagmus. No ptosis.
Parinaud syndrome/dorsal midbrain lesion: Bilateral mid-dilated pupils that react poorly to light but constrict normally with convergence (not tonic). Associated with eyelid retraction (Collier Sign), supranuclear upgaze paralysis, and convergence retraction nystagmus. No ptosis.
 GCA: Extraocular muscle ischemia due to involvement of the long posterior ciliary arteries. Any extraocular muscle may be affected, resulting in potentially complex horizontal and vertical motility deficits. Pupil typically not involved. Age ≥55 years. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
GCA: Extraocular muscle ischemia due to involvement of the long posterior ciliary arteries. Any extraocular muscle may be affected, resulting in potentially complex horizontal and vertical motility deficits. Pupil typically not involved. Age ≥55 years. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
Etiology
 Pupil involving:
Pupil involving:
—More common: Aneurysm, particularly posterior communicating artery aneurysm.
—Less common: Tumor, trauma, congenital, uncal herniation, cavernous sinus mass lesion, pituitary apoplexy, orbital disease, varicella zoster virus, and leukemia. In children, ophthalmoplegic migraine.
 Pupil sparing: Ischemic microvascular disease; rarely cavernous sinus syndrome or GCA.
Pupil sparing: Ischemic microvascular disease; rarely cavernous sinus syndrome or GCA.
 Relative pupil sparing: Ischemic microvascular disease; less likely compressive.
Relative pupil sparing: Ischemic microvascular disease; less likely compressive.
 Aberrant regeneration present: Trauma, aneurysm, tumor, congenital. Not microvascular. See 10.6, Aberrant Regeneration of the Third Nerve.
Aberrant regeneration present: Trauma, aneurysm, tumor, congenital. Not microvascular. See 10.6, Aberrant Regeneration of the Third Nerve.
Work-Up
1. History: Onset and duration of diplopia? Recent trauma? Pertinent medical history [e.g., diabetes, hypertension, known cancer or central nervous system (CNS) mass, recent infections]. If ≥55 years old, specifically ask about GCA symptoms.
2. Complete ocular examination: Check for pupillary involvement, the directions of motility restriction (in both eyes), ptosis, a visual field defect (visual fields by confrontation), proptosis, resistance to retropulsion, orbicularis muscle weakness, and eyelid fatigue with sustained upgaze. Look carefully for signs of aberrant regeneration. See 10.6, Aberrant Regeneration of the Third Nerve.
3. Full neurologic examination: Carefully assess the other cranial nerves on both sides.
NOTE: The ipsilateral fourth nerve can be assessed by focusing on a superior conjunctival blood vessel and asking the patient to look down and nasally. The eye should intort, and the blood vessel should turn down and toward the nose even if the eye cannot be adducted.
4. Immediate CNS imaging to rule out mass/aneurysm is indicated for:
a. Pupil-involving (relatively or completely involved) third nerve palsies.
b. Pupil-sparing third nerve palsies in the following groups of patients:
—Patients ≤50 years of age (unless there is known long-standing diabetes or hypertension).
—Patients with incomplete third nerve palsies (with sparing of some muscle function) because this condition may be evolving into a pupil-involving third nerve palsy. If imaging not obtained, closely monitor patients for pupil involvement daily for at least 7 days.
—Patients with additional cranial nerve or neurologic abnormalities.
c. Children <10 years of age, regardless of the state of the pupil.
NOTE: Most sensitive modality is contrast-enhanced CT and CTA, though gadolinium-enhanced MRI and MRA are also very sensitive and can be done if CT and CTA are contraindicated or unavailable. Choice of imaging should be made in conjunction with neuroradiology. If initial imaging studies are negative but clinical suspicion remains high, catheter angiography may be indicated.
5. Prompt CNS imaging is required for:
a. All patients in whom aberrant regeneration develops, with the exception of regeneration after traumatic third nerve palsies.
b. Pupil-sparing third nerve palsy for >3 months in duration without improvement.
6. Imaging is usually not required in complete pupil-sparing third nerve palsies that do not fit these criteria, especially in patients >50 years of age with known vasculopathic risk factors such as diabetes or hypertension.
7. Cerebral angiography is indicated for all patients >10 years of age with pupil-involving third nerve palsies and whose imaging study is negative or shows a mass consistent with an aneurysm.
8. CBC with differential in children.
9. Edrophonium chloride test or ice-pack test when myasthenia gravis is suspected. See 10.11, Myasthenia Gravis.
10. For suspected ischemic disease: Check blood pressure, fasting blood sugar, glycosylated hemoglobin.
11. Immediate erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and platelets if GCA is suspected. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
Treatment
1. Treat the underlying abnormality.
2. If the third nerve palsy is causing symptomatic diplopia, an occlusion patch or prism may be placed over the involved eye. Patching is usually not performed in children <11 years of age because of the risk of amblyopia. Children should be monitored closely for the development of amblyopia in the deviated eye.
3. Strabismus surgery may be considered for persistent significant misalignment.
Follow-Up
1. Pupil sparing: Observe daily for 7 days from onset of symptoms for delayed pupil involvement, and then recheck every 4 to 6 weeks. If secondary to ischemia, function should return within 3 months. If the palsy does not reverse or is not improving by 3 months, the pupil dilates, additional neurologic abnormalities develop, aberrant regeneration appears, or an incomplete third nerve palsy progresses, then urgent imaging is obtained. Refer to internist for management of vasculopathic disease risk factors.
2. Pupil involving: If imaging and angiography are negative, an LP should be considered and the patient should be followed as described above.
10.6 ABERRANT REGENERATION OF THE THIRD NERVE
Symptoms
See 10.5, Isolated Third Nerve Palsy.
Signs
(See Figures 10.6.1 and 10.6.2.)
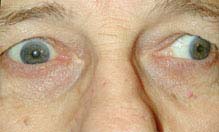
FIGURE 10.6.2. Aberrant regeneration of right third cranial nerve showing right upper eyelid retraction on attempted left gaze.
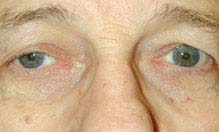
FIGURE 10.6.1. Aberrant regeneration of right third cranial nerve showing right-sided ptosis in primary gaze.
The most common signs of aberrant third nerve regeneration include:
 Eyelid-gaze dyskinesis: Elevation of involved eyelid on downgaze (Pseudo-von Graefe sign) or adduction.
Eyelid-gaze dyskinesis: Elevation of involved eyelid on downgaze (Pseudo-von Graefe sign) or adduction.
 Pupil-gaze dyskinesis: Pupil constricts on downgaze or adduction.
Pupil-gaze dyskinesis: Pupil constricts on downgaze or adduction.
 Other signs may include limitation of elevation and depression of eye, adduction of involved eye on attempted elevation or depression, absent optokinetic response, or pupillary light-near dissociation.
Other signs may include limitation of elevation and depression of eye, adduction of involved eye on attempted elevation or depression, absent optokinetic response, or pupillary light-near dissociation.
Etiology
Thought to result from misdirection of the third nerve fibers from their original destination to alternate third nerve controlled muscles (e.g., inferior rectus to the pupil).
 Aberrancy from congenital third nerve palsies: Can be seen in up to two-thirds of these patients.
Aberrancy from congenital third nerve palsies: Can be seen in up to two-thirds of these patients.
 Aberrancy from prior acquired third nerve palsies: Seen most often in patients recovering from third nerve damage by trauma or compression by a posterior communicating artery aneurysm.
Aberrancy from prior acquired third nerve palsies: Seen most often in patients recovering from third nerve damage by trauma or compression by a posterior communicating artery aneurysm.
 Primary aberrant regeneration: A term used to describe the presence of aberrant regeneration in a patient who has no history of a third nerve palsy. Usually indicates the presence of a progressively enlarging parasellar lesion such as a carotid aneurysm within the cavernous sinus or tumor such as meningioma.
Primary aberrant regeneration: A term used to describe the presence of aberrant regeneration in a patient who has no history of a third nerve palsy. Usually indicates the presence of a progressively enlarging parasellar lesion such as a carotid aneurysm within the cavernous sinus or tumor such as meningioma.
Work-Up
1. Aberrancy from congenital: None. Document work-up of prior congenital third nerve palsy.
2. Aberrancy from acquired: See 10.5, Isolated Third Nerve Palsy. Document work-up of prior acquired third nerve palsy if previously obtained.
3. Primary aberrancy: All patients must undergo neuroimaging to rule out slowly compressive lesion or aneurysm.
NOTE: Ischemic third nerve palsies DO NOT produce aberrancy. If aberrant regeneration develops in a presumed ischemic palsy, neuroimaging should be performed.
Treatment
1. Treat the underlying disorder.
2. Consider strabismus surgery if significant symptoms are present.
Follow-Up
1. Aberrancy from congenital: Routine.
2. Aberrancy from acquired: As per the underlying disorder identified in the work-up.
3. Primary aberrancy: As per neuroimaging findings. Patients are instructed to return immediately for any changes (e.g., ptosis, diplopia, sensory abnormality).
10.7 ISOLATED FOURTH NERVE PALSY
Symptoms
Binocular vertical (or oblique) diplopia, difficulty reading, sensation that objects appear tilted; may be asymptomatic.
Signs
(See Figures 10.7.1 and 10.7.2.)
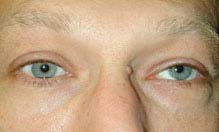
FIGURE 10.7.1. Isolated left fourth cranial nerve palsy: Primary gaze showing left hypertropia.
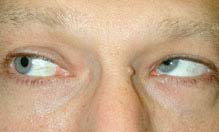
FIGURE 10.7.2. Isolated left fourth cranial nerve palsy: Right gaze with left inferior oblique overaction.
Critical. Deficient inferior movement of an eye when attempting to look down and in. The three-step test isolates a palsy of the superior oblique muscle (see below).
Other. The involved eye is higher (hypertropic) in primary gaze. The hypertropia increases when looking in the direction of the uninvolved eye or tilting the head toward the ipsilateral shoulder. The patient often maintains a head tilt toward the contralateral shoulder to eliminate diplopia.
Differential Diagnosis
All of the following may produce binocular vertical diplopia, hypertropia, or both.
 Myasthenia gravis: Variable symptoms with fatigability. Ptosis common. Orbicularis oculi weakness often present. See 10.11, Myasthenia Gravis.
Myasthenia gravis: Variable symptoms with fatigability. Ptosis common. Orbicularis oculi weakness often present. See 10.11, Myasthenia Gravis.
 Thyroid eye disease: May have proptosis, eyelid lag, lid retraction, or injection over the involved rectus muscles. Positive forced-duction test. See 7.2.1, Thyroid Eye Disease. See Appendix 6, Forced-Duction Test and Active Force Generation Test.
Thyroid eye disease: May have proptosis, eyelid lag, lid retraction, or injection over the involved rectus muscles. Positive forced-duction test. See 7.2.1, Thyroid Eye Disease. See Appendix 6, Forced-Duction Test and Active Force Generation Test.
 Idiopathic orbital inflammatory syndrome: Pain and proptosis are common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
Idiopathic orbital inflammatory syndrome: Pain and proptosis are common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
 Orbital fracture: History of trauma. Positive forced-duction test. See 3.9, Orbital Blow-out Fracture.
Orbital fracture: History of trauma. Positive forced-duction test. See 3.9, Orbital Blow-out Fracture.
 Skew deviation: The three-step test does not isolate a particular muscle. Rule out a posterior fossa or brainstem lesion by MRI of the brain. See 10.13, Internuclear Ophthalmoplegia.
Skew deviation: The three-step test does not isolate a particular muscle. Rule out a posterior fossa or brainstem lesion by MRI of the brain. See 10.13, Internuclear Ophthalmoplegia.
 Incomplete third nerve palsy: Inability to look down and in, usually with adduction weakness. Intorsion on attempted downgaze. Three-step test does not isolate the superior oblique. See 10.5, Isolated Third Nerve Palsy.
Incomplete third nerve palsy: Inability to look down and in, usually with adduction weakness. Intorsion on attempted downgaze. Three-step test does not isolate the superior oblique. See 10.5, Isolated Third Nerve Palsy.
 Brown syndrome: Limitation of elevation in adduction due to restriction of superior oblique tendon. May be congenital or acquired (e.g., trauma, inflammation). Positive forced-duction test. See 8.6, Strabismus Syndromes.
Brown syndrome: Limitation of elevation in adduction due to restriction of superior oblique tendon. May be congenital or acquired (e.g., trauma, inflammation). Positive forced-duction test. See 8.6, Strabismus Syndromes.
 GCA: Extraocular muscle ischemia causing nonspecific motility deficits or neural ischemia mimicking a cranial nerve palsy. Age ≥55 years, usually associated systemic symptoms. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
GCA: Extraocular muscle ischemia causing nonspecific motility deficits or neural ischemia mimicking a cranial nerve palsy. Age ≥55 years, usually associated systemic symptoms. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
Etiology
More common: Trauma, vascular infarct (often the result of underlying diabetes or hypertension), congenital, idiopathic, or demyelinating disease.
Rare: Tumor, hydrocephalus, aneurysm, GCA.
Work-Up
1. History: Onset and duration of the diplopia? Misaligned eyes or head tilt since early childhood? Trauma? Stroke?
2. Examine old photographs to determine whether the head tilt is long standing, indicating an old or congenital fourth nerve palsy.
3. Perform the three-step test:
Step 1: Determine which eye is deviated upward in primary gaze. This is best seen with the cover–uncover test (see Appendix 3, Cover/Uncover and Alternate Cover Tests). The higher eye comes down after being uncovered.
Step 2: Determine whether the upward deviation is greater when the patient looks to the left or to the right.
Step 3: Determine whether the upward deviation is greater when tilting the head to the left shoulder or right shoulder.
Patients with a superior oblique muscle paresis have a hyperdeviation that is worse when turning the elevated eye nasally and when tilting the head toward the shoulder ipsilateral to the elevated eye.
Patients with bilateral fourth nerve palsies demonstrate hypertropia of the right eye when looking left, hypertropia of the left eye when looking right, and a “V”-pattern esotropia (the eyes cross more when looking down).
In addition to the findings on the three-step test, the hypertropia should be greater in downgaze than in upgaze.
4. Perform the double Maddox rod test if bilateral fourth nerve palsies are suspected to measure total excyclotorsion.
—A white Maddox rod is placed before one eye and a red Maddox rod is placed before the other eye in a trial frame or phoropter, aligning the axes of each rod along the 90-degrees vertical mark. While looking at a white light in the distance, the patient is asked if both the white and red lines seen through the Maddox rods are horizontal and parallel to each other. If not, the patient is asked to rotate the Maddox rod(s) until they are parallel. If he or she rotates the top of this vertical axis outward (away from the nose) for more than 10 degrees total for the two eyes, then a bilateral superior oblique muscle paresis is likely present.
5. Measure vertical fusional amplitudes with a vertical prism bar to distinguish a congenital from an acquired palsy.
—A patient with an acquired fourth nerve palsy has a normal vertical fusional amplitude of 1 to 3 prism diopters. A patient with a congenital fourth nerve palsy has >3 prism diopters (sometimes up to 10 to 15 prism diopters) of fusional amplitude.
6. Edrophonium chloride test, ice test, or rest test if myasthenia gravis is suspected (see 10.11, Myasthenia Gravis).
7. CT scan of head and orbits (axial and coronal views) for suspected orbital disease.
8. Blood pressure measurement, fasting blood sugar, and glycosylated hemoglobin. Immediate ESR, CRP, and platelets if GCA is suspected.
9. MRI of the brain for:
—A fourth nerve palsy accompanied by other cranial nerve or neurologic abnormalities.
—All patients <45 years of age with no history of significant head trauma, and patients aged 45 to 55 years with no vasculopathic risk factors or trauma.
Treatment
1. Treat the underlying disorder.
2. An occlusion patch may be placed over one eye or fogging plastic tape can be applied to one lens of patient’s spectacles to relieve symptomatic double vision. Patching is usually not performed in children <11 years of age because of the risk of amblyopia.
3. Prisms in spectacles may be prescribed for small, stable hyperdeviations.
4. Strabismus surgery may be indicated for bothersome double vision in primary or reading position or for a cosmetically significant head tilt. Defer surgery at least 6 months after the onset of the palsy for the deviation to stabilize and because many palsies resolve spontaneously.
Follow-Up
1. Congenital fourth nerve palsy: Routine.
2. Acquired fourth nerve palsy: As per the underlying disorder. If the work-up is negative, the lesion is presumed vascular or idiopathic and the patient is reexamined in 1 to 3 months. If the palsy does not resolve in 3 months or if an additional neurologic abnormality develops, appropriate imaging studies of the brain are indicated. Patients are instructed to return immediately for any changes (e.g., ptosis, worsening diplopia, sensory abnormality, pupil abnormality).
10.8 ISOLATED SIXTH NERVE PALSY
Symptoms
Binocular horizontal diplopia, worse for distance than near, most pronounced in the direction of the paretic lateral rectus muscle.
Signs
(See Figures 10.8.1 and 10.8.2.)
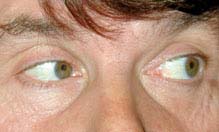
FIGURE 10.8.1. Isolated right sixth cranial nerve palsy: Left gaze showing full adduction.
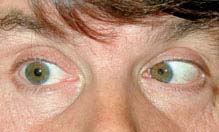
FIGURE 10.8.2. Isolated right sixth cranial nerve palsy: Right gaze showing limited abduction.
Critical. Deficient lateral movement of an eye with negative forced-duction testing (see Appendix 6, Forced-Duction Test and Active Force Generation Test).
Other. No proptosis.
Differential Diagnosis of Limited Abduction
 Thyroid eye disease: May have proptosis, eyelid lag, lid retraction, injection over the involved rectus muscles, and positive forced-duction testing. See 7.2.1, Thyroid Eye Disease.
Thyroid eye disease: May have proptosis, eyelid lag, lid retraction, injection over the involved rectus muscles, and positive forced-duction testing. See 7.2.1, Thyroid Eye Disease.
 Myasthenia gravis: Variable symptoms with fatigability. Ptosis common. Positive edrophonium chloride test, ice test, and rest test. See 10.11, Myasthenia Gravis.
Myasthenia gravis: Variable symptoms with fatigability. Ptosis common. Positive edrophonium chloride test, ice test, and rest test. See 10.11, Myasthenia Gravis.
 Idiopathic orbital inflammatory syndrome: Pain and proptosis are common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
Idiopathic orbital inflammatory syndrome: Pain and proptosis are common. See 7.2.2, Idiopathic Orbital Inflammatory Syndrome.
 Orbital trauma: Fracture causing medial rectus entrapment, positive forced-duction testing. See 3.9, Orbital Blow-out Fracture.
Orbital trauma: Fracture causing medial rectus entrapment, positive forced-duction testing. See 3.9, Orbital Blow-out Fracture.
 Duane syndrome, type 1: Congenital; narrowing of the palpebral fissure and retraction of the globe on adduction. See 8.6, Strabismus Syndromes.
Duane syndrome, type 1: Congenital; narrowing of the palpebral fissure and retraction of the globe on adduction. See 8.6, Strabismus Syndromes.
 Möbius syndrome: Congenital; bilateral facial paralysis present. See 8.6, Strabismus Syndromes.
Möbius syndrome: Congenital; bilateral facial paralysis present. See 8.6, Strabismus Syndromes.
 Convergence spasm: The pupils constrict on attempted abduction.
Convergence spasm: The pupils constrict on attempted abduction.
 Primary divergence insufficiency: Usually acquired and benign; esotropia and diplopia only at distance and single binocular vision at near. Symptoms may improve spontaneously without treatment or may be corrected with base-out prisms or surgery. If the history reveals sudden onset, trauma, infection (e.g., meningitis, encephalitis), multiple sclerosis (MS), or malignancy, divergence paralysis should be considered and a neurologic work-up with MRI of the brain and brainstem obtained.
Primary divergence insufficiency: Usually acquired and benign; esotropia and diplopia only at distance and single binocular vision at near. Symptoms may improve spontaneously without treatment or may be corrected with base-out prisms or surgery. If the history reveals sudden onset, trauma, infection (e.g., meningitis, encephalitis), multiple sclerosis (MS), or malignancy, divergence paralysis should be considered and a neurologic work-up with MRI of the brain and brainstem obtained.
 GCA: Extraocular muscle ischemia, age ≥55 years may be associated with systemic symptoms. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
GCA: Extraocular muscle ischemia, age ≥55 years may be associated with systemic symptoms. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
Etiology
Adults
More common: Vasculopathic (e.g., diabetes, hypertension, other atherosclerotic risk factors), trauma, idiopathic.
Less common: Increased intracranial pressure, cavernous sinus mass (e.g., meningioma, aneurysm, metastasis), MS, sarcoidosis, vasculitis, after myelography or LP, stroke (usually with other neurologic deficits), meningeal inflammation/infection (e.g., Lyme disease, neurosyphilis), GCA.
Children
Benign and usually self-limited after viral infection or vaccination, trauma, increased intracranial pressure (e.g., obstructive hydrocephalus), pontine glioma, Gradenigo syndrome (petrositis causing sixth and often seventh nerve involvement, with or without eighth and fifth nerve involvement on the same side; associated with complicated otitis media).
Work-Up
Adults
1. History: Do the symptoms fluctuate during the day? Cancer, diabetes, or thyroid disease? Symptoms of GCA (in the appropriate age group)?
2. Complete neurologic and ophthalmic examinations; pay careful attention to the function of the other cranial nerves and the appearance of the optic disc. Because of the risk of corneal damage, it is especially important to evaluate the fifth cranial nerve. Corneal sensation (supplied by the first division) can be tested by touching a wisp of cotton or a tissue to the corneas before applying topical anesthetic. Ophthalmoscopy looking for papilledema is required because increased intracranial pressure from any cause can result in unilateral or bilateral sixth nerve palsies.
3. Check blood pressure, fasting blood sugar, and glycosylated hemoglobin.
4. MRI of the brain is indicated for the following patients:
—Younger than 45 years of age (if MRI is negative, consider LP).
—Consider MRI for patients aged 45 to 55 years with no vasculopathic risk factors.
—Sixth nerve palsy accompanied by severe pain or any other neurologic or neuroophthalmic signs.
—Any history of cancer.
—Bilateral sixth nerve palsies.
—Papilledema is present.
5. Immediate ESR, CRP, and platelet count if GCA is suspected. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
6. Consider RPR, FTA-ABS, Lyme titer.
Children
1. History: Recent illness or trauma? Neurologic symptoms, lethargy, or behavioral changes? Chronic ear infections?
2. Complete neurologic and ophthalmic examinations as described for adults.
3. Otoscopic examination to rule out complicated otitis media.
4. MRI of the brain in all children.
Treatment
1. Treat any underlying problem revealed by the work-up.
2. An occlusion patch may be placed over one eye or fogging plastic tape applied to one spectacle lens to relieve symptomatic diplopia. In patients <11 years, patching is avoided, and these patients are monitored closely for the development of amblyopia. See 8.7, Amblyopia.
3. Prisms in glasses may be fit acutely for temporary relief or for chronic stable deviations (e.g., after stroke). Consider strabismus surgery for a stable deviation that persists >6 months.
Follow-Up
Reexamine every 6 weeks after the onset of the palsy until it resolves. MRI of the head is indicated if any new neurologic signs or symptoms develop, the abduction deficit increases, or the isolated sixth nerve palsy does not resolve in 3 to 6 months.
10.9 ISOLATED SEVENTH NERVE PALSY
Symptoms
Weakness or paralysis of one side of the face, inability to close one eye, excessive drooling.
Signs
(See Figures 10.9.1 and 10.9.2.)
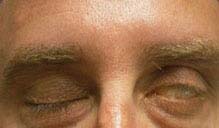
FIGURE 10.9.1. Isolated left seventh cranial nerve palsy: Lagophthalmos.
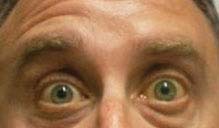
FIGURE 10.9.2. Isolated peripheral left seventh cranial nerve palsy.
Critical. Unilateral weakness or paralysis of the facial musculature.
—Central lesion: Weakness or paralysis of lower facial musculature only. Upper eyelid closure and forehead wrinkling intact.
—Peripheral lesion: Weakness or paralysis of upper and lower facial musculature.
Other. Flattened nasolabial fold, droop of corner of the mouth, ectropion, and lagophthalmos. May have ipsilateral decreased taste on anterior two-thirds of tongue, decreased basic tear production, or hyperacusis. May have an injected eye with a corneal epithelial defect. Synkinesis, a simultaneous movement of muscles supplied by different branches of the facial nerve or simultaneous stimulation of visceral efferent fibers of facial nerve [e.g., corner of mouth contracts when eye closes, excessive lacrimation when eating (“crocodile” tears)], secondary to aberrant regeneration implying chronicity.
Etiology
Central Lesions
 Cortical: Lesion of contralateral motor cortex or internal capsule (e.g., stroke, tumor). Loss of voluntary facial movement; emotional facial movement sometimes intact. May also have ipsilateral hemiparesis.
Cortical: Lesion of contralateral motor cortex or internal capsule (e.g., stroke, tumor). Loss of voluntary facial movement; emotional facial movement sometimes intact. May also have ipsilateral hemiparesis.
 Extrapyramidal: Lesion of basal ganglia (e.g., parkinsonism, tumor, vascular lesion of basal ganglia). Loss of emotional facial movement; volitional facial movement intact. Not a true facial paralysis.
Extrapyramidal: Lesion of basal ganglia (e.g., parkinsonism, tumor, vascular lesion of basal ganglia). Loss of emotional facial movement; volitional facial movement intact. Not a true facial paralysis.
 Brainstem: Lesion of ipsilateral pons (e.g., MS, stroke, tumor). Often with ipsilateral sixth nerve palsy, contralateral hemiparesis. Occasionally with cerebellar signs.
Brainstem: Lesion of ipsilateral pons (e.g., MS, stroke, tumor). Often with ipsilateral sixth nerve palsy, contralateral hemiparesis. Occasionally with cerebellar signs.
Peripheral Lesions
 Cerebellopontine angle (CPA) masses (e.g., acoustic neuroma, facial neuroma, meningioma, cholesteatoma, metastasis): Gradual progressive onset, although sometimes acute. May have facial pain, twitching, or a characteristic nystagmus. This is small-amplitude rapid jerk nystagmus in which the fast phase is directed away from the side of the lesion (peripheral vestibular) in conjunction with a slow, gaze-evoked nystagmus directed toward the side of the lesion (from brainstem compression). May have eighth nerve dysfunction, including hearing loss, tinnitus, vertigo, or dysequilibrium.
Cerebellopontine angle (CPA) masses (e.g., acoustic neuroma, facial neuroma, meningioma, cholesteatoma, metastasis): Gradual progressive onset, although sometimes acute. May have facial pain, twitching, or a characteristic nystagmus. This is small-amplitude rapid jerk nystagmus in which the fast phase is directed away from the side of the lesion (peripheral vestibular) in conjunction with a slow, gaze-evoked nystagmus directed toward the side of the lesion (from brainstem compression). May have eighth nerve dysfunction, including hearing loss, tinnitus, vertigo, or dysequilibrium.
 Temporal bone fracture: History of head trauma. May have Battle sign (ecchymoses over mastoid region), cerebrospinal fluid otorrhea, hearing loss, vertigo, or vestibular nystagmus.
Temporal bone fracture: History of head trauma. May have Battle sign (ecchymoses over mastoid region), cerebrospinal fluid otorrhea, hearing loss, vertigo, or vestibular nystagmus.
 Other trauma: Accidental or iatrogenic (e.g., facial laceration, local anesthetic block, parotid or mastoid surgery).
Other trauma: Accidental or iatrogenic (e.g., facial laceration, local anesthetic block, parotid or mastoid surgery).
 Acute or chronic suppurative otitis media.
Acute or chronic suppurative otitis media.
 Malignant otitis externa: Pseudomonas infection in diabetic or elderly patients. Begins in external auditory canal but may progress to osteomyelitis, meningitis, or abscess.
Malignant otitis externa: Pseudomonas infection in diabetic or elderly patients. Begins in external auditory canal but may progress to osteomyelitis, meningitis, or abscess.
 Ramsay–Hunt syndrome (varicella zoster oticus): Viral prodrome followed by ear pain; vesicles on pinna, external auditory canal, tongue, face, or neck. Progresses over 10 days. May have sensorineural hearing loss, tinnitus, or vertigo.
Ramsay–Hunt syndrome (varicella zoster oticus): Viral prodrome followed by ear pain; vesicles on pinna, external auditory canal, tongue, face, or neck. Progresses over 10 days. May have sensorineural hearing loss, tinnitus, or vertigo.
 Guillain–Barré syndrome: Viral syndrome followed by progressive motor weakness or paralysis or cranial nerve palsies, or both. Loss of deep tendon reflexes. May have bilateral facial palsies.
Guillain–Barré syndrome: Viral syndrome followed by progressive motor weakness or paralysis or cranial nerve palsies, or both. Loss of deep tendon reflexes. May have bilateral facial palsies.
 Lyme disease: May have rash, fever, fatigue, arthralgias, myalgias, or nausea. There may or may not be a history of tick bite. See 13.3, Lyme Disease.
Lyme disease: May have rash, fever, fatigue, arthralgias, myalgias, or nausea. There may or may not be a history of tick bite. See 13.3, Lyme Disease.
 Sarcoidosis: May have uveitis, parotitis, skin lesions, or lymphadenopathy. May have bilateral facial palsies. See 12.6, Sarcoidosis.
Sarcoidosis: May have uveitis, parotitis, skin lesions, or lymphadenopathy. May have bilateral facial palsies. See 12.6, Sarcoidosis.
 Parotid neoplasm: Slowly progressive paralysis of all or portion of facial musculature. Parotid mass with facial pain.
Parotid neoplasm: Slowly progressive paralysis of all or portion of facial musculature. Parotid mass with facial pain.
 Metastasis: History of primary tumor (e.g., breast, lung, prostate). Multiple cranial nerve palsies in rapid succession may be seen. Can be the result of basilar skull metastasis or carcinomatous meningitis.
Metastasis: History of primary tumor (e.g., breast, lung, prostate). Multiple cranial nerve palsies in rapid succession may be seen. Can be the result of basilar skull metastasis or carcinomatous meningitis.
 Bell palsy: Idiopathic seventh nerve palsy. Most common, but other etiologies must be ruled out. May have viral prodrome followed by ear pain, facial numbness, decreased tearing or taste. Facial palsy may be complete or incomplete and progress over 10 days. May be recurrent, rarely bilateral. Possible familial predisposition.
Bell palsy: Idiopathic seventh nerve palsy. Most common, but other etiologies must be ruled out. May have viral prodrome followed by ear pain, facial numbness, decreased tearing or taste. Facial palsy may be complete or incomplete and progress over 10 days. May be recurrent, rarely bilateral. Possible familial predisposition.
 Others: Diabetes mellitus, botulism, human immunodeficiency virus (HIV), syphilis, Epstein–Barr virus, acute porphyrias, nasopharyngeal carcinoma, collagen–vascular disease, and others.
Others: Diabetes mellitus, botulism, human immunodeficiency virus (HIV), syphilis, Epstein–Barr virus, acute porphyrias, nasopharyngeal carcinoma, collagen–vascular disease, and others.
Work-Up
1. History: Onset and duration of facial weakness? First episode or recurrence? Facial or ear pain? Trauma? Stroke? Recent infection? Hearing loss, tinnitus, dizziness, or vertigo? History of sarcoidosis or cancer?
2. Examine old photographs to determine chronicity of facial droop.
3. Complete neurologic examination: Determine if facial palsy is central or peripheral, complete or incomplete. Assess taste with bitter or sweet solution on anterior two-thirds of tongue on affected side. Carefully assess other cranial nerves, especially the fifth, sixth, and seventh. Look for motor weakness and cerebellar signs.
4. Complete ocular examination: Check ocular motility and look for nystagmus. Assess orbicularis strength bilaterally, degree of ectropion, and Bell phenomenon. Examine cornea carefully for signs of exposure (superficial punctate keratopathy, epithelial defect, or ulcer). Perform Schirmer test (see 4.3, Dry-Eye Syndrome) to assess basic tear production. Check for signs of uveitis.
5. Otolaryngologic examination: Examine ear and oropharynx for vesicles, masses, or other lesions. Palpate parotid for mass or lymphadenopathy. Check hearing.
6. CT scan if history of trauma to rule out basilar skull fracture: Axial and coronal cuts with attention to temporal bone.
7. MRI or CT scan of brain if any other associated neurologic signs, history of cancer, or duration >3 months. Sixth nerve involvement warrants attention to the brainstem. Eighth nerve involvement warrants attention to the CPA. Multiple cranial nerve involvement warrants attention to the skull base.
8. Chest radiograph and angiotensin-
converting enzyme (ACE) level if sarcoidosis suspected.
9. Consider Lyme titer, Epstein–Barr virus titer, RPR, HIV test, and CBC with differential, depending on suspected etiology.
10. Rheumatoid factor, ESR, antinuclear antibody (ANA), and antineutrophil cytoplasmic antibody if collagen–vascular disease suspected.
11. Echocardiogram, Holter monitor, carotid noninvasive studies in patients with a history of stroke.
12. LP in patients with history of primary neoplasm to rule out carcinomatous meningitis (repeat up to three times if negative to increase sensitivity).
Treatment
1. Treat the underlying disease as follows:
—Stroke: Refer to neurologist.
—CPA masses, temporal bone fracture, nerve laceration: Refer to neurosurgeon.
—Otitis: Refer to otolaryngologist.
—Ramsay–Hunt syndrome: If seen within 72 hours of onset, start acyclovir 800 mg five times per day for 7 to 10 days (contraindicated in pregnancy and renal failure). Refer to otolaryngologist.
—Guillain–Barré syndrome: Refer to neurologist. May require urgent hospitalization for rapidly progressive motor weakness or respiratory distress.
—Lyme disease: Refer to infectious disease specialist. May need LP. Treat with oral doxycycline, penicillin, or intravenous (i.v.) ceftriaxone. See 13.3, Lyme Disease.
—Sarcoidosis: Treat uveitis if present. Consider brain MRI, LP, or both to rule out CNS involvement; if present, refer to neurologist. Refer to internist for systemic evaluation. May require systemic prednisone for extraocular or CNS disease. See 12.6, Sarcoidosis.
—Metastatic disease: Refer to oncologist. Systemic chemotherapy, radiation, or both may be required.
2. Bell palsy: 86% of patients recover completely with observation alone within 2 months. Options for treatment include:
—Facial massage or electrical stimulation of facial musculature.
—Consider steroids (e.g., prednisone 60 mg p.o. q.d. for 4 days, tapering to 5 mg q.d. over 10 days). A short course of acyclovir or famciclovir, in combination with prednisone, may improve facial nerve function outcomes.
—Consider referral to otorhinolaryngology for surgical decompression of the facial nerve.
3. The primary ocular complication of facial palsy is corneal exposure, which is managed as follows (also see 4.5, Exposure Keratopathy):
—Mild exposure keratitis: Artificial tears q.i.d. with lubricating ointment q.h.s.
—Moderate exposure keratitis: Preservative-free artificial tears q1–2h, moisture chamber during the day with lubricating ointment, or tape tarsorrhaphy q.h.s. Consider a temporary tarsorrhaphy.
—Severe exposure keratitis: Temporary or permanent tarsorrhaphy. For expected chronic facial palsy, consider eyelid gold weight to facilitate eyelid closure.
Follow-Up
1. Recheck all patients at 1 and 3 months and more frequently if corneal complications arise.
2. If not resolved after 3 months, order MRI of brain to rule out mass lesion.
3. In nonresolving facial palsy with repeatedly negative work-up, consider referral to neurosurgeon or plastic surgeon for facial nerve graft, cranial nerve reanastomosis, or temporalis muscle transposition for patients who strongly desire facial reanimation.
10.10 CAVERNOUS SINUS AND ASSOCIATED SYNDROMES (MULTIPLE OCULAR MOTOR NERVE PALSIES)
Symptoms
Double vision, eyelid droop, facial pain, or numbness.
Signs
Critical. Limitation of eye movement corresponding to any combination of a third, fourth, or sixth nerve palsy on one side; facial pain or numbness or both corresponding to one or more branches of the fifth cranial nerve; ptosis and a small pupil (Horner syndrome); the pupil also may be dilated if the third cranial nerve is involved. Any combination of the above may be present simultaneously because of the anatomy of the cavernous sinus. All signs involve the same side of the face when one cavernous sinus/superior orbital fissure is involved. The circular sinus connects the cavernous sinuses, and its involvement can cause contralateral signs. Consider orbital apex syndrome when proptosis and optic neuropathy are present.
Other. Proptosis may be present when the superior orbital fissure is involved.
Differential Diagnosis
 Myasthenia gravis: Fatigable ptosis, orbicularis weakness, and limited motility. Pupils uninvolved. No proptosis. See 10.11, Myasthenia Gravis.
Myasthenia gravis: Fatigable ptosis, orbicularis weakness, and limited motility. Pupils uninvolved. No proptosis. See 10.11, Myasthenia Gravis.
 CPEO: Progressive, painless, bilateral motility limitation with ptosis. Normal pupils. Orbicularis always weak. See 10.12, Chronic Progressive External Ophthalmoplegia.
CPEO: Progressive, painless, bilateral motility limitation with ptosis. Normal pupils. Orbicularis always weak. See 10.12, Chronic Progressive External Ophthalmoplegia.
 Orbital lesions (e.g., tumor, thyroid disease, inflammation). Proptosis and increased resistance to retropulsion are usually present, in addition to motility restriction. Results of forced-duction tests are abnormal (see Appendix 6, Forced-Duction Test and Active Force Generation Test). May have an afferent pupillary defect if the optic nerve is involved.
Orbital lesions (e.g., tumor, thyroid disease, inflammation). Proptosis and increased resistance to retropulsion are usually present, in addition to motility restriction. Results of forced-duction tests are abnormal (see Appendix 6, Forced-Duction Test and Active Force Generation Test). May have an afferent pupillary defect if the optic nerve is involved.
NOTE: Orbital apex syndrome combines the superior orbital fissure syndrome with optic nerve dysfunction, and most commonly results from an orbital lesion.
 Brainstem disease: Tumors and vascular lesions of the brainstem can produce ocular motor nerve palsies, particularly the sixth cranial nerve. MRI of the brain is best for making this diagnosis.
Brainstem disease: Tumors and vascular lesions of the brainstem can produce ocular motor nerve palsies, particularly the sixth cranial nerve. MRI of the brain is best for making this diagnosis.
 Carcinomatous meningitis: Diffuse seeding of the leptomeninges by metastatic tumor cells can produce a rapidly sequential bilateral cranial nerve disorder. Diagnosis is made by serial LPs.
Carcinomatous meningitis: Diffuse seeding of the leptomeninges by metastatic tumor cells can produce a rapidly sequential bilateral cranial nerve disorder. Diagnosis is made by serial LPs.
 Skull base tumors, especially nasopharyngeal carcinoma or clivus lesions: Most commonly affects the sixth cranial nerve, but the second, third, fourth, and fifth cranial nerves may be involved as well. Typically, one cranial nerve after another is affected by invasion of the base of the skull. The patient may have cervical lymphadenopathy, nasal obstruction, ear pain, or popping caused by serous otitis media or blockage of the Eustachian tube, weight loss, or proptosis.
Skull base tumors, especially nasopharyngeal carcinoma or clivus lesions: Most commonly affects the sixth cranial nerve, but the second, third, fourth, and fifth cranial nerves may be involved as well. Typically, one cranial nerve after another is affected by invasion of the base of the skull. The patient may have cervical lymphadenopathy, nasal obstruction, ear pain, or popping caused by serous otitis media or blockage of the Eustachian tube, weight loss, or proptosis.
 Progressive supranuclear palsy (PSP): Vertical limitation of eye movements, typically beginning with downward gaze. Postural instability, dementia, and rigidity of the neck and trunk may be present. All eye movements are eventually lost. See 10.12, Chronic Progressive External Ophthalmoplegia.
Progressive supranuclear palsy (PSP): Vertical limitation of eye movements, typically beginning with downward gaze. Postural instability, dementia, and rigidity of the neck and trunk may be present. All eye movements are eventually lost. See 10.12, Chronic Progressive External Ophthalmoplegia.
 Rare: Myotonic dystrophy, bulbar variant of the Guillain–Barré syndrome (Miller–Fisher variant), intracranial sarcoidosis, others.
Rare: Myotonic dystrophy, bulbar variant of the Guillain–Barré syndrome (Miller–Fisher variant), intracranial sarcoidosis, others.
Etiology
 Arteriovenous fistula [carotid–cavernous (“high-flow”) or dural–cavernous (“low-flow”)]: Proptosis, chemosis, dilated and tortuous (“corkscrew”) episcleral and conjunctival blood vessels (see Figure 10.10.1). Intraocular pressure is often increased. Enhanced ocular pulsation (“pulsatile proptosis”) may be present, usually only discernible on slit-lamp examination during applanation. A bruit may be heard by the patient, and sometimes by the physician if the globe or temple region is auscultated. Reversed, arterialized flow in the superior ophthalmic vein is detectable with orbital color Doppler US. Orbital CT scan or MRI may show an enlarged superior ophthalmic vein. High-flow fistulas have an abrupt onset and are most commonly caused by trauma or rupture of an intracavernous aneurysm, whereas low-flow fistulas have a more insidious presentation, most commonly in hypertensive women >50 years of age and are due to dural arteriovenous malformations. The Barrow classification is used for preoperative planning and further subdivides carotid–cavernous fistulas as follows:
Arteriovenous fistula [carotid–cavernous (“high-flow”) or dural–cavernous (“low-flow”)]: Proptosis, chemosis, dilated and tortuous (“corkscrew”) episcleral and conjunctival blood vessels (see Figure 10.10.1). Intraocular pressure is often increased. Enhanced ocular pulsation (“pulsatile proptosis”) may be present, usually only discernible on slit-lamp examination during applanation. A bruit may be heard by the patient, and sometimes by the physician if the globe or temple region is auscultated. Reversed, arterialized flow in the superior ophthalmic vein is detectable with orbital color Doppler US. Orbital CT scan or MRI may show an enlarged superior ophthalmic vein. High-flow fistulas have an abrupt onset and are most commonly caused by trauma or rupture of an intracavernous aneurysm, whereas low-flow fistulas have a more insidious presentation, most commonly in hypertensive women >50 years of age and are due to dural arteriovenous malformations. The Barrow classification is used for preoperative planning and further subdivides carotid–cavernous fistulas as follows:

FIGURE 10.10.1. Carotid–cavernous fistula with dilated and tortuous episcleral and conjunctival vessels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


