Neuro-Ophthalmology: Introduction
As demonstrated by their common embryological origin, the retinas and anterior visual pathways (optic nerves, optic chiasm, and optic tracts) are an integral part of the brain, providing a substantial proportion of total sensory input. They frequently give important diagnostic clues to central nervous system disorders. Intracranial disease frequently causes visual disturbances because of destruction of or pressure upon some portion of the optic pathways. Cranial nerves III, IV, and VI, which control ocular movements, may be involved, and nerves V and VII are also intimately associated with ocular function.
Cranial nerve II subserves the special sense of vision. Light is detected by the rods and cones of the retina, which may be considered the special sensory end organ for vision. The cell bodies of these receptors extend processes that synapse with the bipolar cell, the second neuron in the visual pathway. The bipolar cells synapse, in turn, with the retinal ganglion cells. Ganglion cell axons comprise the nerve fiber layer of the retina and converge to form the optic nerve. The nerve emerges from the back of the globe and travels posteriorly within the muscle cone to enter the cranial cavity via the optic canal.

Intracranially, the two optic nerves join to form the optic chiasm (Figure 14–1). At the chiasm, more than half of the fibers (those from the nasal half of the retina) decussate and join the uncrossed temporal fibers of the opposite nerve to form the optic tracts. Each optic tract sweeps around the cerebral peduncle toward the lateral geniculate nucleus, where it will synapse. All of the fibers receiving impulses from the right hemifields of each eye thus make up the left optic tract and project to the left cerebral hemisphere. Similarly, the left hemifields project to the right cerebral hemisphere. Twenty percent of the fibers in the tract subserve pupillary function. These fibers leave the tract just anterior to the nucleus and pass via the brachium of the superior colliculus to the midbrain pretectal nucleus. The remaining fibers synapse in the lateral geniculate nucleus. The cell bodies of this structure give rise to the geniculocalcarine tract. This tract passes through the posterior limb of the internal capsule and then fans into the optic radiations that traverse parts of the temporal and parietal lobes en route to the occipital (calcarine, striate, or primary visual) cortex.
In clinical practice, lesions in the visual pathways may be localized by examination of the central and peripheral visual fields. The technique (perimetry) is discussed in Chapter 2.
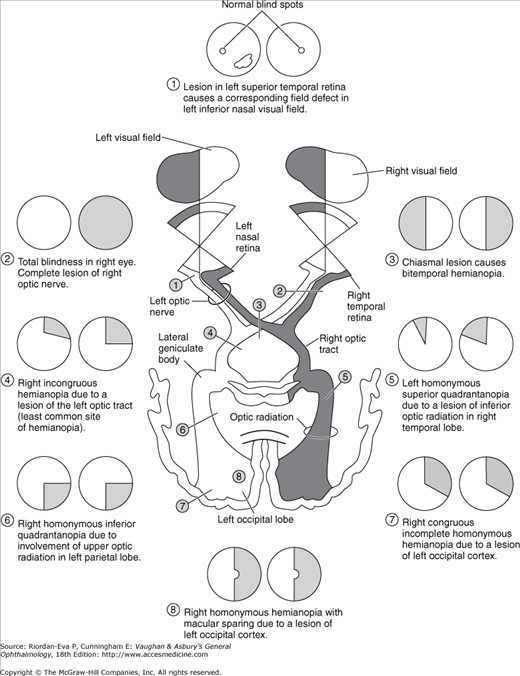
Figure 14–3 shows the types of field defects caused by lesions in various locations of the pathway. Lesions anterior to the chiasm (of the retina or optic nerve) cause unilateral field defects; lesions anywhere in the visual pathway posterior to the chiasm cause contralateral homonymous defects (affecting the same side of the visual field in each eye). Chiasmal lesions usually cause bitemporal defects.
Test objects of different intensities, sizes, and possibly colors are used to evaluate field defects thoroughly. A sloping border (ie, larger field defect to a dimmer, smaller or colored rather than white test object) suggests edema or compression. Ischemic or vascular lesions tend to produce steep borders (ie, the field defect is the same size no matter what the intensity, size, or color of the test object).
The more congruous the homonymous field defects (ie, the more similar the defect in size, shape, and location in the two eyes), the farther posterior the lesion is likely to be in the visual pathway. A lesion in the occipital region tends to cause identical defects in each field, whereas optic tract lesions tend to cause incongruous (dissimilar) homonymous field defects.
Particularly in lesions of the occipital cortex, where the central field is represented posteriorly and the upper field inferiorly (Figure 14–4), there is a correlation between the visual field defect and the location of the lesion. Owing to the dual vascular supply to the occipital lobe—from the middle and posterior cerebral circulation—occipital infarcts may spare or damage the occipital pole. This leads to sparing or loss of the central field on the side of the hemianopia, respectively referred to as macular sparing (Figure 14–5) and macular splitting. Occipital lesions may also produce the phenomenon of residual sight, in which responses to movement, for example, may be demonstrable in the hemianopic field in the absence of form vision.
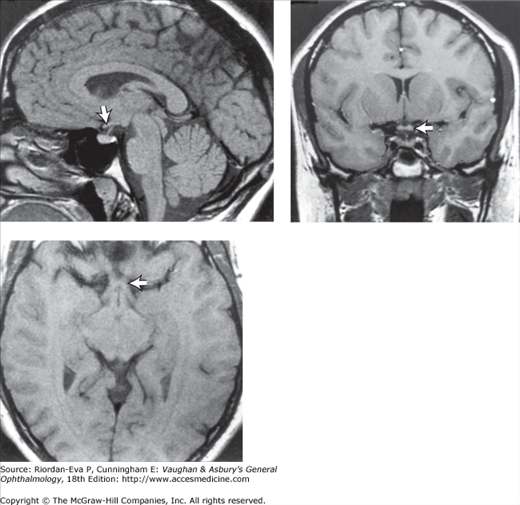
Figure 14-4.

Occipital lobe abscess. Top: Automated perimetry and tangent screen examination showing homonymous, congruous, paracentral scotoma in right upper visual fields. Bottom: Parasagittal MRI showing lesion involving left inferior calcarine cortex. (Reproduced, with permission, from Horton JC et al: The representation of the visual field in human striate cortex. A revision of the classic Holmes map. Arch Ophthalmol 1991;109:816.)
Figure 14-5.
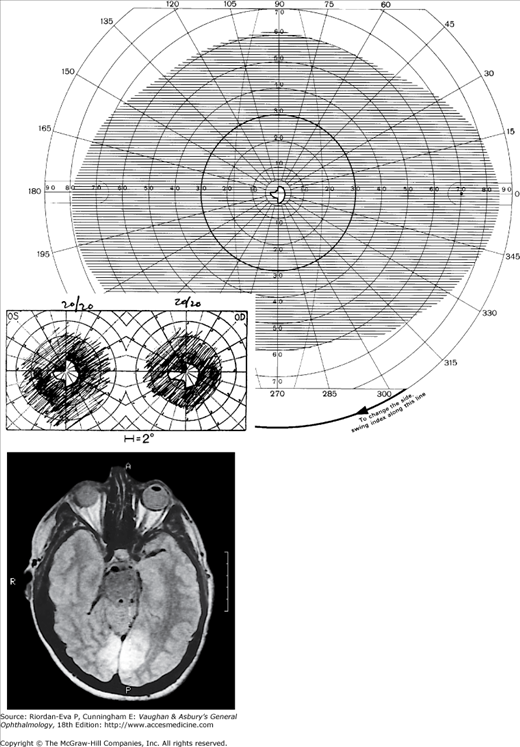
Bilateral occipital infarcts with bilateral macular sparing. Top: Tangent screen and superimposed Goldmann visual fields of both eyes showing bilateral homonymous hemianopia with macular sparing, greater in the right hemi-field. Bottom: Axial MRI showing sparing of occipital poles. (Reproduced, with permission, from Horton JC et al: The representation of the visual field in human striate cortex. A revision of the classic Holmes map. Arch Ophthalmol 1991;109:816.)
A complete homonymous hemianopia, wherever the site of the lesion, should still have intact visual acuity in each eye since macular function is still present in the retained visual field.
The Optic Nerve
A wide variety of diseases affect the optic nerve (Table 14–1). Clinical features particularly suggestive of optic nerve disease are an afferent pupillary defect, poor color vision, and optic disk changes. It is important to remember that the optic disk may be normal in the early stages of disease affecting the retrobulbar optic nerve, particularly compression by an intracranial lesion, even when there has been severe loss of visual acuity and field. Axons can be dysfunctional long before they become atrophic.
| Inflammatory (optic neuritis) |
| Demyelinative |
| Multiple sclerosis |
| Neuromyelitis optica (Devic’s disease) |
| Postviral (measles, mumps, chickenpox, influenza, infectious mononucleosis) |
| Postimmunization |
| Acute disseminated encephalomyelitis |
| Immune-mediated |
| Systemic lupus erythematosus |
| Granulomatous |
| Sarcoidosis |
| Direct infection |
| Herpes zoster, syphillis, tuberculosis, cryptococcosis, cytomegalovirus, Lyme disease, cat-scratch disease |
| Contiguous inflammatory disease |
| Intraocular inflammation |
| Orbital disease |
| Sinus disease, including mucormycosis |
| Intracranial disease: meningitis, encephalitis |
| Vascular (ischemic optic neuropathy) |
| Nonarteritic anterior ischemic optic neuropathy |
| Systemic vasculitis |
| Giant cell arteritis (arteritic anterior ischemic optic neuropathy), systemic lupus erythematosus, polyarteritis nodosa |
| Diabetic papillopathy |
| Posterior ischemic optic neuropathy |
| Sudden massive blood loss (eg, trauma, gastrointestinal hemorrhage) |
| Radiation optic neuropathy |
| Raised intracranial pressure (papilledema) |
| Intracranial mass: cerebral tumor, abscess, subdural hematoma |
| Arteriovenous malformation |
| Subarachnoid hemorrhage |
| Meningitis or encephalitis |
| Hydrocephalus |
| Pseudotumor cerebri |
| Cerebral or jugular venous sinus occlusion |
| Secondary pseudotumor cerebri: oral contraceptives, tetracyclines, steroid therapy, steroid withdrawal, hypervitaminosis A, uremia, hypoparathyroidism, obstructive sleep apnea, respiratory failure |
| Idiopathic intracranial hypertension |
| Spinal tumor |
| Acute idiopathic polyneuropathy (Guillain-Barré syndrome) |
| Mucopolysaccharidosis |
| Craniosynostosis |
| Optic nerve compression |
| Intracranial disease: meningioma, pituitary adenoma, craniopharyngioma, supraclinoid internal carotid aneurysm, meningeal carcinomatosis, basal meningitis |
| Orbital disease: dysthyroid eye disease, idiopathic orbital inflammatory disease, orbital neoplasm, orbital abscess |
| Optic nerve sheath meningioma |
| Nutritional and toxic |
| Vitamin deficiencies: vitamin B12, vitamin B1 (thiamin), folate |
| Tobacco-alcohol amblyopia |
| Heavy metals: lead, thallium, arsenic |
| Drugs: ethambutol, isoniazid, linezolid, disulfiram, tamoxifen, quinine, chloramphenicol, amiodarone, chemotherapeutic agents |
| Chemicals: methanol, ethylene glycol |
| Trauma |
| Direct optic nerve injury |
| Indirect optic nerve injury |
| Optic nerve avulsion |
| Hereditary optic atrophy |
| Leber’s hereditary optic neuropathy (mitochondrial inheritance) |
| Autosomal hereditary optic atrophy |
| Dominant (juvenile) optic atrophy |
| Recessive (infantile) optic atrophy |
| Wolfram’s syndrome (DIDMOAD: diabetes insipidus, diabetes mellitus, optic atrophy, deafness) |
| Inherited neurodegenerative diseases |
| Hereditary spinocerebellar ataxia (Friedreich’s ataxia) |
| Hereditary motor and sensory neuropathy (Charcot-Marie-Tooth disease) |
| Lysosomal storage disorders |
| Neoplastic infiltration |
| Glioma, leukemia, lymphoma, meningeal carcinomatosis, astrocytic hamartoma, melanocytoma, hemangioma |
| Optic nerve anomalies |
| Hypoplasia |
| Dysplasia (including “morning glory syndrome,” coloboma, and optic nerve pit) |
| Tilted disks, including situs inversus, and scleral crescents |
| Megalopapilla |
| Myelinated nerve fibers |
| Persistent hyaloid system |
| Prepapillary vascular loops |
| Optic nerve head drusen |
| Hyperopic pseudopapilledema |
| Glaucomatous optic neuropathy (see Chapter 11: Glaucoma) |
| Optic atrophy secondary to retinal disease |
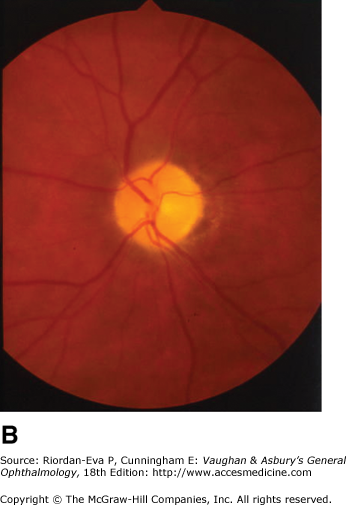
Optic disk swelling occurs predominantly in diseases directly affecting the anterior portion of the optic nerve but also occurs with raised intracranial pressure and compression of the intraorbital optic nerve. Optic disk swelling can be a crucial clinical sign, such as in the diagnosis of anterior ischemic optic neuropathy, in which optic disk swelling must be present in the acute stage for the diagnosis to be made on clinical grounds. Central retinal vein occlusion, ocular hypotony, and intraocular inflammation can produce optic disk swelling, and hence the misleading impression of optic nerve disease.
Optic atrophy (Figure 14–6) is a nonspecific response to optic nerve damage from any cause. Since the optic nerve consists of retinal ganglion cell axons, optic atrophy may be the consequence of primary retinal disease, such as central retinal artery occlusion or retinitis pigmentosa. Excavation of the optic nerve head (optic disk cupping) is generally a sign of glaucomatous optic neuropathy, but it may occur with any cause of optic atrophy. Segmental pallor and attenuated retinal blood vessels are often the consequence of anterior ischemic optic neuropathy. Hereditary optic neuropathies usually produce bilateral temporal segmental disk pallor with preferential loss of papillomacular axons. Peripapillary exudates occur with optic disk swelling, due to inflammation (papillitis), ischemic optic neuropathy, raised intracranial pressure (papilloedema), or severe systemic hypertension. (The term “neuroretinitis” for retinal exudates, including a macular star, due to optic disk swelling, of whatever cause, is a misnomer in that there is no inflammation of the retina, the exudates being a response to the anterior optic nerve disease (Figure 14–7 A). The term is more reasonably applied if there is inflammation of the retina and optic nerve (Figure 14–7 B). Signs of prior disk edema are peripapillary gliosis and atrophy, chorioretinal folds, and internal limiting membrane wrinkling.
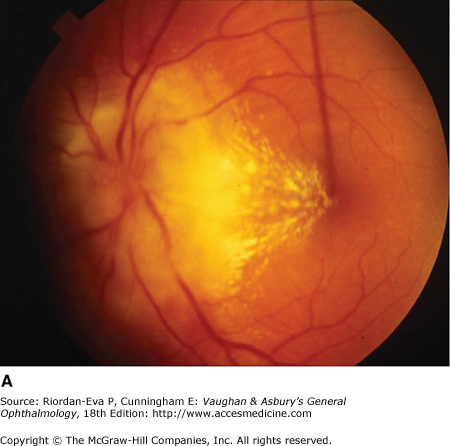
Figure 14-6.
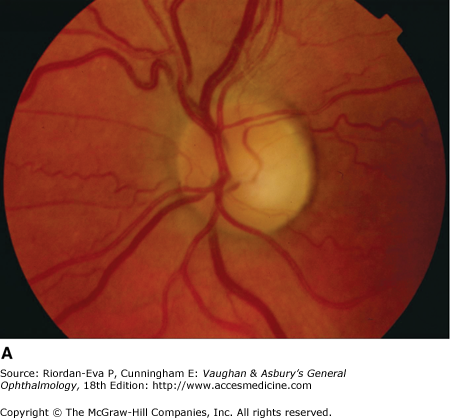
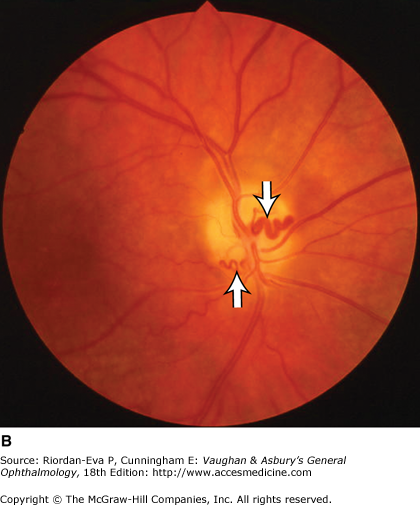
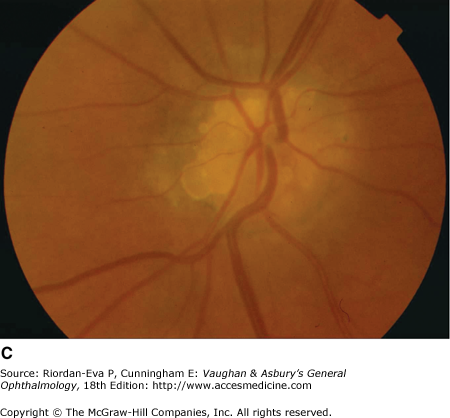
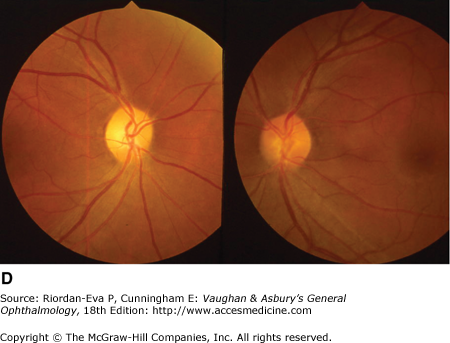
Examples of optic atrophy. A: Primary optic atrophy due to nutritional amblyopia. B: Secondary optic atrophy with retinochoroidal collaterals (arrows) due to optic nerve sheath meningioma. C: Optic atrophy with optic disk drusen. D: Pallor (atrophy) of right optic disk due to nerve compression by sphenoid meningioma. The left disk is normal.
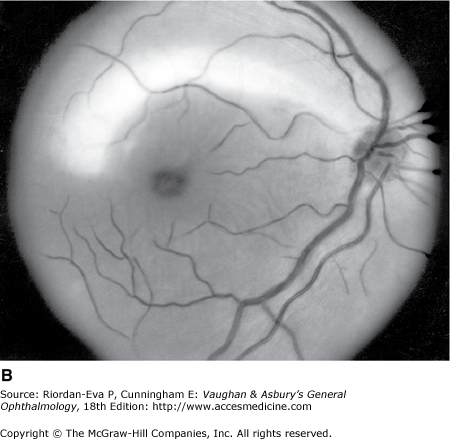
In general, there is a correlation between degree of optic disk pallor and loss of acuity, visual field, color vision, and pupillary responses, but the relationship varies according to the underlying etiology. The major exception to this rule is compressive optic neuropathy, in which optic disk pallor is generally a late manifestation.
Inflammatory optic neuropathy (optic neuritis) may be due to a variety of causes (Table 14–1), but the most common is demyelinative disease, including multiple sclerosis. Retrobulbar neuritis is an optic neuritis that occurs far enough behind the optic disk that the disk remains normal during the acute episode. Papillitis is disk swelling caused by inflammation at the nerve head (intraocular optic nerve) (Figure 14–8). Loss of vision is the cardinal symptom of optic neuritis and is particularly useful in differentiating papillitis from papilledema, with which it may be confused on ophthalmoscopic examination.

In adults demyelinative optic neuritis is usually unilateral and occurs chiefly in women (about 3:1), with onset mostly in the third or fourth decade of life. It is associated with multiple sclerosis in up to 85% of cases, depending on several factors, including gender, racial origin, and duration of follow-up. In children there is much lower risk of progression to multiple sclerosis, optic neuritis commonly following a viral infection or immunization and affecting both eyes simultaneously.
Visual loss is generally subacute, developing over 2–7 days. In adults, approximately one-third of patients have vision better than 20/40 during their first attack, and slightly more than one-third have vision worse than 20/200. Color vision and contrast sensitivity are correspondingly impaired. In over 90% of cases, there is pain in the region of the eye, and about 50% of patients report that the pain is exacerbated by eye movement.
Almost any field defect is possible. With manual perimetry a central scotoma is most commonly found. Central visual field testing by automated perimetry most commonly shows diffuse loss. The pupillary light response is sluggish, and if the optic nerves are asymmetrically involved, a relative afferent pupillary defect will be present.
Optic disk swelling (papillitis) occurs in one third of adult cases and two thirds of childhood cases. Marked edema is unusual. Peripapillary flame-shaped hemorrhages occur in less than 10% of adult cases and vitreous cells in less than 5%. Retinal exudates and cotton-wool spots don’t occur and are indicative of an infectious inflammatory or non-inflammatory optic neuropathy.
Without treatment vision usually improves, visual acuity being better than 20/30 within 2 weeks in the majority of adult cases.
In typical cases, clinical diagnosis is adequate and no other investigation is required. If there are atypical features—particularly failure of vision to begin to recover by 6 weeks after onset—other diagnoses must be considered, especially compressive optic neuropathy, for which magnetic resonance imaging (MRI) or computed tomography (CT) should be performed. Other entities to be considered are optic neuritis due to sarcoidosis or systemic lupus erythematosus, anterior ischemic optic neuropathy, Leber’s hereditary optic neuropathy, toxic amblyopia, and vitamin B12 deficiency, of which the last two usually present with symmetrical bilateral visual loss.
Papillitis needs to be differentiated from papilledema (optic disk swelling due to raised intracranial pressure) (Figure 14–9). In papilledema, which usually causes bilateral optic disk changes, there is often greater elevation of the optic nerve head, normal corrected visual acuity, normal pupillary response to light, and an intact visual field except for an enlarged blind spot. If there has been acute papilledema with vascular decompensation (ie, hemorrhages and cotton-wool spots) or chronic papilledema with secondary ischemia of the optic nerve, visual field defects can include nerve fiber bundle defects and nasal quadrantanopias. Differentiation between papilledema and papillitis is particularly difficult when papilledema is asymmetric and/or associated with visual loss or papillitis is bilateral and/or associated with minimal visual impairment. Diagnosis may depend on associated symptoms particularly headache and results of MRI and lumbar puncture, as well as subsequent clinical course.

During an acute episode of optic neuritis, MRI shows gadolinium enhancement, increased signal, and sometimes swelling of the affected nerve. The gadolinium enhancement and optic nerve swelling resolve, but the increased signal persists. Brain MRI at presentation shows white matter lesions in about 50% of patients with isolated optic neuritis (Figure 14–10). This does not establish a diagnosis of multiple sclerosis, although it does indicate an increased risk of subsequent development of clinically definite multiple sclerosis (see later in the chapter).
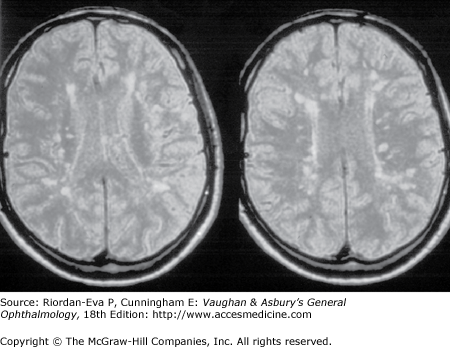
The visual evoked response (VER) from the affected eye may show reduced amplitude or increased latency during the acute episode of optic neuritis. This in itself is not particularly helpful in diagnosis except in distinguishing retrobulbar optic neuritis from subclinical maculopathy, in which there will be abnormalities of the electroretinogram (ERG). Following recovery of vision after an episode of optic neuritis, the VER will continue to show an increased latency in about one-third of cases, and this finding can be useful in the identification of past episodes of demyelinative optic neuritis in patients undergoing investigation for possible multiple sclerosis.
Steroid therapy by whatever route—either intravenously (methylprednisolone, 1 g/d for 3 days with or without a subsequent tapering course of oral prednisolone), orally (methylprednisolone, 500 mg/d to 2 g/d for 3–5 days with or without subsequent oral prednisolone, or prednisolone, 1 mg/kg/d tapered over 10–21 days), or by retrobulbar injection—accelerates recovery of vision but does not influence the ultimate visual outcome. In the large multicenter Optic Neuritis Treatment Trial from the United States, oral prednisolone alone increased the risk of recurrent optic neuritis in either eye, but this result has not been replicated.
Vision continues to improve slowly over many months, with recovery of acuity to 20/40 or better occurring in over 90% of cases at both 1 year and 15 years from onset. Poorer vision during the acute episode is correlated with poorer visual outcome, but even loss of all perception of light can be followed by recovery of acuity to 20/20. Poor visual outcome is also associated with longer lesions in the optic nerve, especially if there is involvement of the nerve within the optic canal. In general, there is close correlation between recovery of visual acuity, contrast sensitivity, and color vision. If the disease process is sufficiently destructive, retrograde optic atrophy results, nerve fiber bundle defects appear in the retinal nerve fiber layer (Figure 14–11), and the disk becomes pale. Factors that correlate with subsequent development of multiple sclerosis include female sex, lack of optic disk swelling during the acute phase, brain MRI abnormalities, and cerebrospinal fluid oligoclonal bands. In the Optic Neuritis Treatment Trial, the overall 15-year risk of developing clinically definite multiple sclerosis following a first episode of demyelinative optic neuritis was 50%. It ranged from 25% in those with normal brain MRI at presentation to 72% in those with any white matter lesions. In men with optic disk swelling in the acute phase and no brain MRI abnormality, the 15-year risk was only 4%. Neurologic impairment in patients who develop clinically definite multiple sclerosis is generally mild and MRI abnormalities may be predictive.

In patients with a first episode of optic neuritis, which is designated a clinically isolated syndrome, and abnormal brain MRI at presentation, interferon β and glatiramer acetate have been shown to reduce the risk of clinically definite multiple sclerosis. Whether patients should be treated with such disease modifying therapy after a first episode of optic neuritis needs to be determined on an individual basis, according to their likely risk of subsequent episodes of demyelination and bearing in mind the relatively mild likely disability after further episodes and the potential side effects of treatment. The benefit is likely to increase if the patient develops frequent relapses.
Multiple sclerosis is typically a chronic relapsing and remitting demyelinating disorder of the central nervous system. The cause is unknown. Some patients develop a chronically progressive form of the disease, either following a period of relapses and remissions (secondary progressive) or, less commonly, from the outset (primary progressive). Characteristically, the lesions occur at different times and in noncontiguous locations in the nervous system—that is, “lesions are disseminated in time and space.” Onset is usually in young adult life; the disease rarely beginning before 15 years or after 55 years of age. There is a tendency to involve the optic nerves and chiasm, brainstem, cerebellar peduncles, and spinal cord, although no part of the central nervous system is protected. The peripheral nervous system is seldom involved.
Optic neuritis may be the first manifestation. There may be recurrent episodes, and the other eye usually becomes involved. The overall incidence of optic neuritis in multiple sclerosis is 90%, and the identification of symptomatic or subclinical optic nerve involvement is an important diagnostic clue.
Diplopia is a common early symptom, due most frequently to internuclear ophthalmoplegia that is frequently bilateral (Figure 14–12). Less common causes are lesions of the sixth or third cranial nerve within the brainstem.

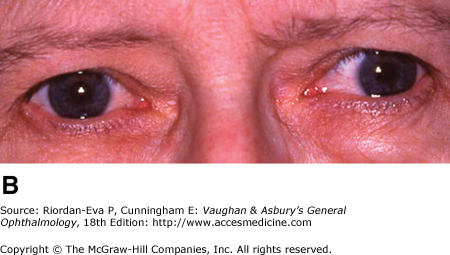
Nystagmus is a common early sign, and unlike most manifestations of the disease (which tend toward remission), it is often permanent (70%).
Intraocular inflammation may occur, particularly subclinical peripheral retinal venous sheathing, which can be highlighted by fluorescein angiography.
In addition to ocular disturbances, there may be motor weakness with pyramidal signs, ataxia, limb incoordination with intention tremor, dysarthria, urinary and/or bowel disturbance, and sensory disturbance, particularly paresthesias.
Diagnosis of multiple sclerosis traditionally relied upon clinical evidence of white matter disease of the central nervous system disseminated in time and space (Schumacher criteria), subsequently supported by MRI and cerebrospinal fluid abnormalities (Poser criteria). Increasingly, emphasis is being placed on MRI abnormalities, in the brain and spinal cord, supported by clinical features and cerebrospinal fluid abnormalities, to establish dissemination in time and space (McDonald criteria), thus facilitating earlier diagnosis.
Cerebrospinal fluid oligoclonal bands that are not present in the serum—representing intrathecal production of immunoglobulins—are characteristic but not diagnostic. There may be cerebrospinal fluid lymphocytosis or a mildly raised cerebrospinal fluid protein concentration during an acute relapse.
Retinal nerve fiber layer defects consistent with a subclinical optic neuritis can be detected in 68% of multiple sclerosis patients. The VER may help confirm involvement of the visual pathway. It has been reported to be abnormal in 80% of definite, 43% of probable, and 22% of suspected cases of multiple sclerosis.
The course of disease is unpredictable. Optic neuritis rather than brainstem or spinal cord disease as the initial manifestation is associated with a better prognosis. Relapses and remissions are characteristic, permanent disability tending to increase with each relapse. Pregnancy or the number of pregnancies has no effect on disability, but there is an increased risk of relapse just after delivery. Onset during pregnancy has a more favorable outcome than onset unrelated to pregnancy. Elevation of body temperature may exacerbate disability (Uhthoff’s phenomenon), particularly visual impairment.
Steroid treatment, usually oral or intravenous methylprednisolone, is useful in hastening recovery from acute relapses but does not influence the final disability or the frequency of subsequent relapses. Interferon β glatiramer acetate, mitoxantrone, and natalizumab reduce the rate and severity of relapses and slow the progression of brain MRI abnormalities but the effect on long-term disability is still being determined. Many immunosuppressant treatments have been tested for progressive disease with no significant benefit.
Neuromyelitis optica (Devic’s disease) is characterized by recurrent optic neuritis and transverse myelitis that may resemble severe multiple sclerosis, but lesions on brain MRI are atypical for multiple sclerosis; spinal cord lesions are long and necrotic; there may be a cellular cerebrospinal fluid response; severe disability is common; and there is a specific probably pathogenic serum auto-antibody, NMO-IgG. The clinical outcome is variable. Approximately 50% of patients progress to death within the first decade due to the paraplegia, but the remainder may have a prolonged remission and, ultimately, a better prognosis than patients with multiple sclerosis. Treatment is with systemic steroids or if necessary plasmapheresis for the acute episodes, followed by long-term immunosuppression, primarily targeted at humoral immunity, according to disease activity.
Particularly in children, 1–2 weeks following a viral infection or immunization there may be an episode of optic neuritis, often with simultaneous bilateral involvement. There is no association with subsequent development of multiple sclerosis. In some cases, the acute disease causes more extensive neurologic involvement manifesting as an encephalomyelitis, which overlaps with acute disseminated encephalomyelitis, in which optic neuritis is also often bilateral.
Optic neuropathy in systemic lupus erythematosus may be immune-mediated, with features of inflammatory disease, or due to small blood vessel occlusion, with features of ischemic disease (see later in the chapter). Inflammatory optic neuropathy may occur in sarcoidosis, sometimes as the first manifestation. Generally there is rapid response to steroid therapy, but long-term treatment with steroids and/or other immunosuppressants is often required. Such disease occurring in individuals in whom no evidence of sarcoidosis or other systemic disease can be identified has been termed idiopathic granulomatous optic neuropathy or chronic relapsing inflammatory optic neuropathy (CRION).
Herpes zoster—particularly herpes zoster ophthalmicus—may be complicated by optic neuropathy. This is probably due to vasculitis as well as direct neural invasion, and the prognosis is poor even with antiviral and steroid therapy. Other types of primary infection of the optic nerve, such as by syphilis, tuberculosis, cryptococcosis, and cytomegalovirus, are becoming more common with the increasing numbers of severely immunocompromised individuals such as those with AIDS. Lyme disease and cat-scratch disease are important causes of optic neuritis associated with macular star formation.
Intraocular inflammation may lead to direct involvement of the anterior optic nerve with visual loss or to optic disk swelling without apparent reduction in optic nerve function. Optic nerve involvement is an important cause of permanent visual loss in cellulitis or vasculitis of the orbit. The association between sinusitis and optic neuritis is less strong than once thought, but the occurrence of visual loss in the presence of sphenoid or posterior ethmoid sinus disease may indicate a causal relationship, particularly if there is a sinus mucocele. In diabetic or immunocompromised patients, mucormycosis is an important cause of rapidly progressive sinus disease with optic and other cranial nerve involvement (see Chapter 13).
Anterior ischemic optic neuropathy is characterized by pallid disk swelling associated with acute loss of vision; often there are one or two peripapillary splinter hemorrhages (Figure 14–13). The disorder is due to infarction of the retrolaminar optic nerve (the region just posterior to the lamina cribrosa) from occlusion or decreased perfusion of the short posterior ciliary arteries. Fluorescein angiography in the acute stage shows decreased perfusion of the optic disk, often segmental in the nonarteritic form but usually diffuse in the arteritic form and disk leakage in the late phase. There may be associated perfusion defects in the peripapillary choroid.
Figure 14-13.
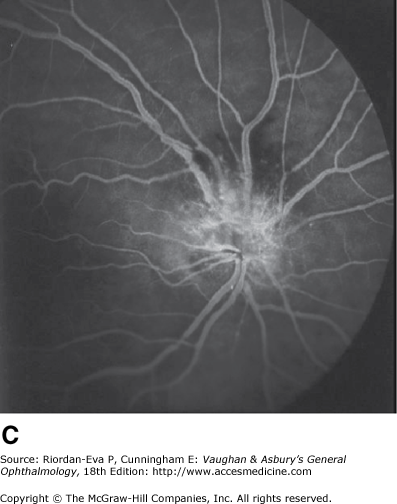
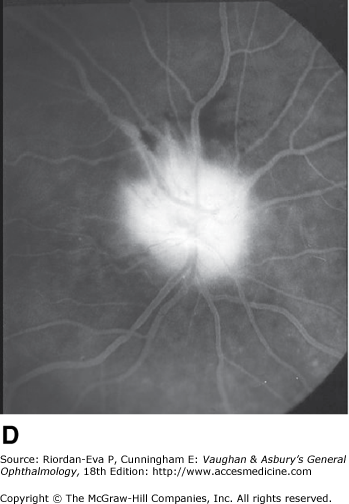
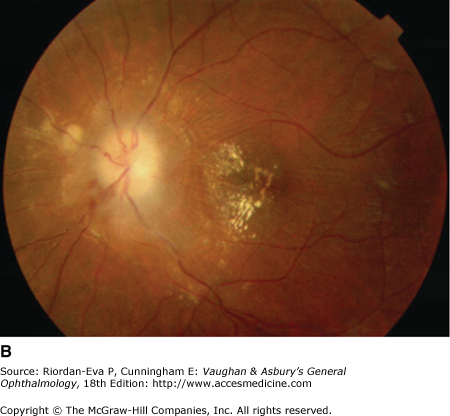
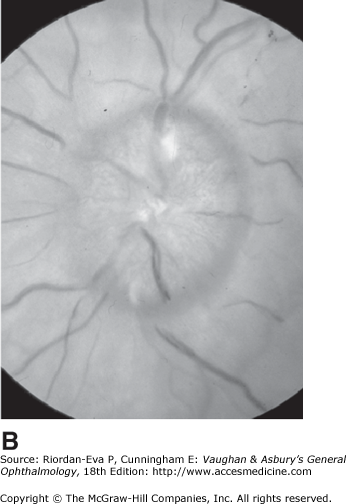
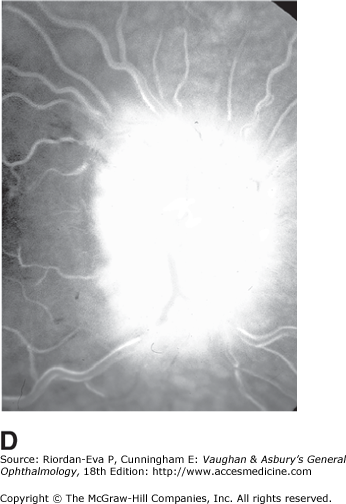
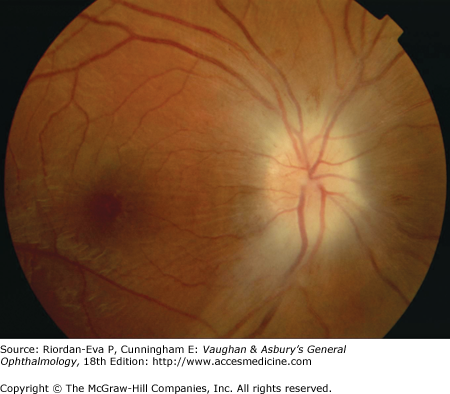
Pseudo-Foster Kennedy syndrome due to sequential anterior ischemic optic neuropathy. A: Swollen right optic disk with hemorrhages due to current ischemic episode. B: Atrophy of left optic disk due to previous ischemia. C: Early phase of fluorescein angiogram of right eye showing poor perfusion of optic disk and dilated superficial disk capillaries. D: Late phase of fluorescein angiogram showing disk leakage.
Nonarteritic anterior ischemic optic neuropathy occurs generally in the sixth or seventh decade and is associated with arteriosclerosis, diabetes, hypertension, and hyperlipidemia. Low optic cup to disk ratio is almost always present. Optic nerve head drusen and increased intraocular pressure are predisposing factors. Systemic hypotension during the early morning may be an important etiologic factor. The precise relationship between phosphodiesterase inhibitors for erectile dysfunction and ischemic optic neuropathy is uncertain. In younger patients, vasculitis (eg, systemic lupus erythematosus, antiphospholipid antibody syndrome, and polyarteritis nodosa), migraine, and inherited prothrombotic states (deficiencies of protein C, protein S, or antithrombin III and activated protein C resistance) should be considered and appropriately treated. The visual loss is generally abrupt, but it may be progressive over 1–2 weeks. Impairment of visual acuity varies from slight to no light perception; visual field defects are usually nasal (characteristically inferior with a relative altitudinal pattern). In over 40% of cases, there is spontaneous improvement in visual acuity. No treatment has been shown to provide long-term benefit. Low-dose aspirin therapy may reduce the risk of involvement of the fellow eye, which occurs in up to 40% of individuals. Recurrences in the same eye are rare. As the acute process resolves, a pale disk usually without “glaucomatous” cupping results.
It is particularly important to identify arteritic anterior ischemic optic neuropathy due to giant cell arteritis. This causes severe visual loss with the risk of complete blindness if treatment is delayed. It occurs in elderly people and is associated with painful and tender temporal arteries, pain on mastication (jaw claudication), general malaise, and muscular aches and pains (polymyalgia rheumatica). The diagnosis is usually based on an anterior ischemic optic neuropathy and high erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) in an elderly patient, with or without associated local or systemic features, but the ESR and CRP may be normal. Other ocular manifestations of giant cell arteritis are central retinal artery occlusion, cilioretinal artery occlusion, retinal cotton-wool spots, ophthalmic artery occlusion, and diffuse ocular ischemia. Diagnosis is established by temporal artery biopsy, looking particularly for inflammatory cell infiltration, often but not always including giant cells, and prominent disruption of the internal elastic lamina.
Treatment with high-dose systemic steroids should be started as soon as a clinical diagnosis of arteritic anterior ischemic optic neuropathy has been made without waiting for the result of temporal artery biopsy, which should be performed within 1 week after starting treatment. Oral prednisolone, 80–100 mg/d, is usually adequate as a starting dose. Intravenous hydrocortisone, 250–500 mg, can be given if there is likely to be a delay in instituting oral therapy. Intravenous methylprednisolone may improve final visual outcome and certainly should be considered in patients with bilateral disease—including those with transient episodes of visual loss in the second eye—and in patients whose visual loss progresses or whose systemic manifestations and high ESR do not respond despite oral therapy. Steroid dosage can usually be reduced to about 40 mg prednisolone per day over 4 weeks but then should be more gradually tapered and discontinued after 9–12 months overall as long as there has been no recurrence of disease activity. Thirty percent of patients require long-term steroid therapy.
Diabetics occasionally develop mild, chronic, usually bilateral disk swelling with little change in visual function, so-called diabetic papillopathy. This is thought to represent microvascular disease affecting the optic disk circulation. It is sometimes confused with optic disk neovascularization because of the leakage of dye from the disk on fluorescein angiography. Posterior ischemic optic neuropathy, in which there is no optic disk swelling during the acute stage of the disease, may occur from massive blood loss, such as from trauma or bleeding peptic ulcer, nonocular surgery, particularly lumbar spine surgery in the prone position, radiotherapy, usually treatment for skull base or sinus tumors 12–18 months previously, giant cell arteritis, or mucormycosis. In general, the diagnosis of posterior ischemic optic neuropathy should not be considered until other causes, particularly a compressive lesion, have been excluded. Radiation optic neuropathy produces a characteristic pattern of gadolinium enhancement on MRI and may be helped by early hyperbaric oxygen therapy.
Papilledema is by definition optic disk swelling due to raised intracranial pressure, of which the most common causes are cerebral tumors, abscesses, subdural hematoma, arteriovenous malformations, subarachnoid hemorrhage, hydrocephalus, meningitis, and encephalitis.
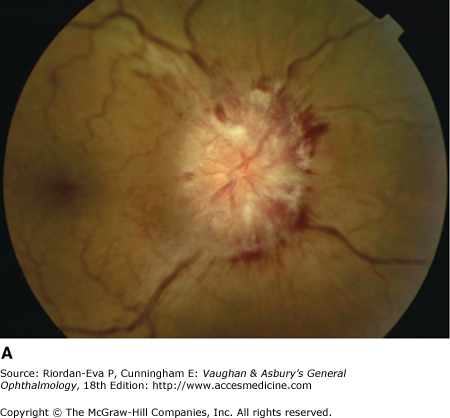


In ophthalmology practice, a frequent cause is idiopathic intracranial hypertension. This is characterized by raised intracranial pressure, no neurologic or neuroimaging abnormality except for anything attributable to the raised intracranial pressure, such as sixth cranial nerve palsy, and normal cerebrospinal fluid constituents. It is a diagnosis of exclusion, and a number of other causes of the syndrome of pseudotumor cerebri must be excluded, such as cerebral venous sinus occlusion, tetracycline or vitamin A (retinoid) therapy, and particularly in men, obstructive sleep apnea.
Less common causes of papilledema are spinal tumors, acute idiopathic polyneuropathy (Guillain–Barré syndrome), mucopolysaccharidosis, and craniosynostoses, in which various factors, including decreased cerebrospinal fluid absorption, abnormalities of spinal fluid flow, and reduced cranial volume, contribute to the raised intracranial pressure.
For papilledema to occur, the subarachnoid space around the optic nerve must be patent and connect the retrolaminar optic nerve through the bony optic canal to the intracranial subarachnoid space, thus allowing increased intracranial pressure to be transmitted to the retrolaminar optic nerve. There, slow and fast axonal transport is blocked, and axonal distention, particularly noticeable at the superior and inferior poles of the optic disk, occurs as the first sign of papilledema. Hyperemia of the disk with dilated surface capillaries, blurring of the peripapillary disk margin, and loss of spontaneous venous pulsations are the signs of mild papilledema. Circumferential peripapillary retinal folds (Paton’s lines) also develop. In acute papilledema, probably as a consequence either of markedly elevated or rapidly increasing intracranial pressure, there are hemorrhages and cotton-wool spots on and around the optic disk, indicating vascular and axonal decompensation with the attendant risk of acute optic nerve damage and visual field defects (Figure 14–14). There may also be peripapillary edema (which can extend to the macula), retinal exudates (Figure 14–14), and choroidal folds. In chronic papilledema (Figure 14–15), which is likely to be the consequence of prolonged, moderately raised intracranial pressure, a process of compensation appears to limit the optic disk changes such that there are few if any hemorrhages or cotton-wool spots. With persistent raised intracranial pressure, the hyperemic elevated disk gradually becomes gray-white as a result of astrocytic gliosis and neural atrophy with secondary constriction of retinal blood vessels, thus leading to the stage of atrophic papilledema (Figure 14–16). There may also be retinochoroidal collaterals (previously known as opticociliary shunts) linking the central retinal vein and the peripapillary choroidal veins, which develop when the retinal venous circulation is obstructed in the prelaminar region of the optic nerve. (Other causes of retinochoroidal collaterals are central retinal vein occlusion, optic nerve sheath meningioma (Figure 14–6), optic nerve glioma, and optic nerve head drusen.) The presence of drusen-like deposits within the swollen optic nerve head, which indicates that the swelling is likely to have been present for several months, characterizes vintage papilledema.
It takes 24–48 hours for early papilledema to occur and 1 week to develop fully. It takes 6–8 weeks for fully developed papilledema to resolve following adequate treatment. Transient visual obscurations are a characteristic symptom of papilledema. Acute papilledema may reduce visual acuity by causing hyperopia and occasionally is associated with optic nerve infarction, but in most cases vision is normal apart from blind spot enlargement. Chronic, particularly atrophic or vintage, papilledema is associated with gradual constriction of the peripheral visual field, particularly inferonasal loss. Sudden reduction of intracranial pressure or systolic perfusion pressure may precipitate severe visual loss in any stage of papilledema.
Papilledema is often asymmetric. It may even appear to be unilateral, although fluorescein angiography in such cases usually shows leakage from both disks. Papilledema occurs late in glaucoma and will not occur at all if there is optic atrophy or if the optic nerve sheath on that side is not patent. The Foster Kennedy syndrome is papilledema on one side with optic atrophy due to optic nerve compression on the other, commonly due to skull-base meningiomas. However, it can be mimicked (pseudo-Foster Kennedy syndrome) by ischemic optic neuropathy when optic disk swelling due to a new episode of ischemic optic neuropathy is associated with optic atrophy in the fellow eye due to a previous episode (Figure 14–13).
Papilledema can be mimicked by buried optic nerve head drusen, small hyperopic disks, and myelinated nerve fibers (Figure 14–17).
The treatment of papilledema must be directed to the underlying cause. Idiopathic intracranial hypertension generally affects obese young women, and maintained weight loss is then an important treatment objective. The major morbidity is visual loss due to papilledema, but headaches may also be troublesome. Oral acetazolamide—usually 250 mg one to four times daily but up to 500 mg four times daily in severe cases—or diuretics such as furosemide are usually effective in reducing optic disk swelling. Cerebrospinal fluid shunting or optic nerve sheath fenestration may be undertaken if there is severe or progressive loss of vision or if medical therapy is not tolerated. Repeated lumbar punctures are rarely indicated except as a temporary measure prior to surgical therapy. Headaches usually respond to control of intracranial pressure, but other treatments may be required. It is essential that patients with idiopathic intracranial hypertension undergo regular visual field assessments by perimetry.
Optic nerve compression is often amenable to treatment, and early recognition is vital to optimal outcome. The possibility of optic nerve compression should be considered in any patient with signs of optic neuropathy or visual loss not explained by an intraocular lesion. Optic disk swelling may occur with intraorbital optic nerve compression, but in many cases, particularly when the optic nerve compression is intracranial, the optic disk shows no abnormality until optic atrophy develops or there is papilledema from associated raised intracranial pressure. (Examination for signs of optic nerve disease, particularly a relative afferent pupillary defect, is thus crucial in assessment of the patient with unexplained visual loss.) Investigation of possible optic nerve compression requires early imaging by MRI or CT. If no structural lesion is identified and meningeal disease is suspected, it may be necessary to proceed to cerebrospinal fluid examination.
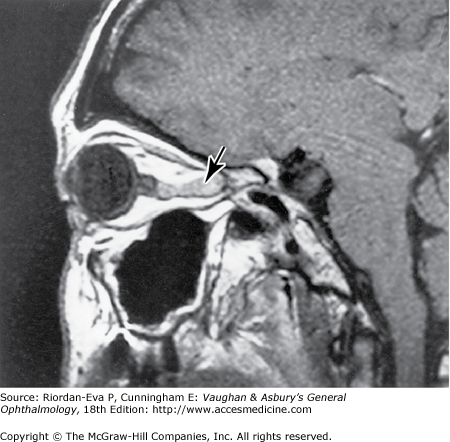
Intracranial meningiomas that may compress the optic nerve include those arising from the sphenoid wing, the tuberculum sellae (suprasellar meningioma), and the olfactory groove. Sphenoid wing meningiomas also produce proptosis, ocular motility disturbance, and trigeminal sensory loss (Figure 14–18). Surgical excision is generally effective in debulking intracranial meningiomas, but complete excision is often very difficult to achieve and recurrence rates are relatively high. Radiotherapy may be indicated as adjuvant or primary treatment. Pituitary adenoma and craniopharyngioma are discussed in the section on chiasmal disease (see later in the chapter). The management of orbital causes of optic nerve compression is discussed in Chapter 13.
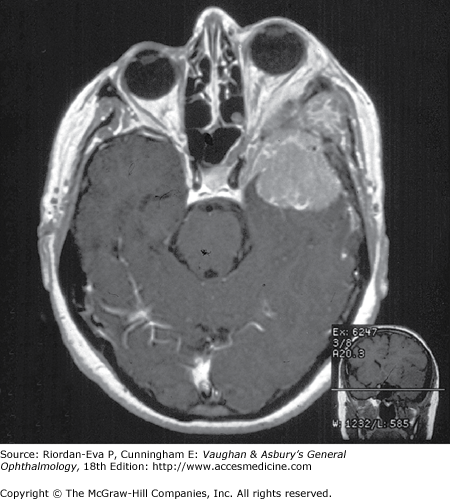
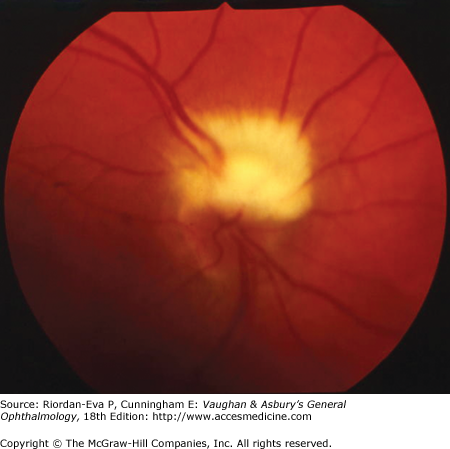
Primary optic nerve sheath meningioma is a rare tumor most commonly presenting, like other types of meningioma, in middle-aged women (Figure 14–19). Five percent of cases are bilateral. Visual loss is slowly progressive. The classic clinical features are a pale, slightly swollen optic disk with retinochoroidal collaterals, but in most cases the collateral vessels are not present (Figure 14–6). Stereotactic radiotherapy is the preferred treatment.
The usual clinical features of nutritional or toxic optic neuropathy are subacute, progressive, symmetrical visual loss, with central field defects (Figure 14–20), poor color vision, and the development of temporal disk pallor (Figure 14–6).

Optic nerve involvement is relatively uncommon in vitamin B12 deficiency, but it may be the first manifestation of pernicious anemia. Thiamin (vitamin B1) deficiency is generally a feature of severe malnutrition, and, as discussed below, there is an overlap with tobacco-alcohol amblyopia. Folate deficiency alone is a rare cause of optic neuropathy.
Nutritional amblyopia is probably a more accurate term for this entity. Usually it occurs in individuals with poor dietary habits, with heavy alcohol consumption and heavy smoking often being associated. Strict vegetarianism without vitamin supplementation may contribute. Much consideration has been given to other toxic causes, such as cyanide from tobacco producing low vitamin stores and low levels of sulfur-containing amino acids, but experimental studies with cyanide in primates have not confirmed this theory. Drug-induced optic neuropathy must be excluded. Leber’s hereditary optic neuropathy, pernicious anemia, methanol poisoning, the rare chronic optic neuropathy of primary progressive multiple sclerosis, or macular disease may cause diagnostic confusion.
There is bilateral loss of central vision, generally reducing visual acuity to less than 20/200, but it can be asymmetric. Central visual fields reveal scotomas that nearly always include both fixation and the blind spot (centrocecal scotoma) (Figure 14–20).
Adequate diet plus thiamine, folic acid, and vitamin B12 supplements may be effective if presentation is not delayed. Withdrawal of tobacco and alcohol is advisable and may hasten the cure, but adequate nutrition or vitamin B12 supplements can be effective despite continued excessive intake of alcohol or tobacco. Improvement usually begins within 1–2 months, although in occasional cases significant improvement may not occur for 1 year. Visual function can but may not return to normal. Permanent optic atrophy or at least temporal disk pallor can occur depending on the stage of disease at the time treatment was started (Figure 14–6). Loss of the ganglion cells of the macula and destruction of myelinated fibers of the optic nerve—and sometimes of the chiasm as well—are the main histologic changes.
Chronic lead exposure, or thallium (present in depilatory cream) or arsenic poisoning can produce a toxic effect on the optic nerve.
Ethambutol, isoniazid, linezolid, disulfiram, and tamoxifen can all cause optic neuropathy, which usually improves with prompt cessation of the drug with or without nutritional supplements. Quinine overdose produces optic neuropathy, narrowed retinal arterioles and irregular, poorly reactive pupils. Chloramphenicol in high doses causes optic neuropathy. Amiodarone has been associated with bilateral optic neuropathy with chronic disk swelling, but the relationship is not necessarily causal. It characteristically also induces a verticillate keratopathy. Various chemotherapeutic agents may cause optic neuropathy, especially with high-dose or intra-arterial therapy.
Absorption, usually oral, of methanol, which is used widely in the chemical industry as antifreeze, solvent varnish, or paint remover, causes visual impairment, sometimes progressing to complete blindness. Whitish, striated edema of the peripapillary retina is a characteristic sign.
Treatment consists of correction of the acidosis with intravenous sodium bicarbonate and oral or intravenous administration of ethanol to compete with, and thus prevent the slower metabolism of methanol into its byproducts. Hemodialysis is indicated for blood methanol levels over 50 mg/dL.
Direct optic nerve injury occurs in penetrating orbital trauma, including local anesthetic injections for ocular surgery, and in mid-facial fractures involving the optic canal. Visual loss due to indirect optic nerve trauma, which refers to optic nerve damage secondary to distant skull injury, occurs in approximately 1% of all head injuries. The site of injury is usually the forehead, often without skull fracture, and the probable mechanism of optic nerve injury is transmission of shock waves through the orbital walls to the orbital apex. Optic nerve avulsion usually results from an abrupt rotational injury to the globe, such as from being poked in the eye with a finger.
Surgery may be indicated to relieve orbital, subperiosteal, or optic nerve sheath hemorrhage or to treat orbital fractures. High-dose systemic steroids for direct or indirect optic nerve injury and decompression of the bony optic canal for indirect injury have been advocated, but their value is uncertain. There is no effective treatment for optic nerve avulsion.
Leber’s hereditary optic neuropathy is a rare disease characterized by sequential subacute optic neuropathy usually in males aged 11–30 years. The underlying genetic abnormality is a point mutation in mitochondrial DNA (mtDNA), with over 90% of affected families harboring a mutation at position 11778, 14484, or 3460. mtDNA is exclusively derived from the mother, and thus, in accordance with the general pattern of mitochondrial (maternal) inheritance (see Chapter 18), the mutation is transmitted through the female line—but for unexplained reasons the disease rarely manifests in carrier females. Once an individual is known to have the disorder, it is possible without further genetic testing to predict which other family members are at risk, with matrilineal nephews, that is, sons of the affected individual’s sisters, being particularly at risk.
Blurred vision and a central scotoma usually appear first in one eye and later—within days, weeks, or months—in the other eye. During the acute episode, there may be swelling of the optic disk and peripapillary retina with dilated telangiectatic small blood vessels on their surface, but characteristically there is no leak from the optic disk during fluorescein angiography. Both optic nerves eventually become atrophic, and vision is usually between 20/200 and counting fingers. The 14484 mutation is associated with recovery of vision but not until many months after the initial onset of visual loss. Total loss of vision or recurrences of visual loss usually do not occur. Leber’s neuropathy may be associated with a multiple sclerosis-like illness (particularly in affected females), cardiac conduction defects, and dystonia.
Diagnosis is by identification of one of the three mtDNA point mutations. No treatment is known to be effective. Because high tobacco and alcohol consumption may precipitate visual loss in susceptible individuals, carriers of a pathogenic point mutation, particularly males, should be advised not to smoke and to avoid high alcohol consumption.
Optic atrophy also occurs in other mitochondrial disorders, either as a manifestation of primary optic neuropathy—for example, myoclonic epilepsy and ragged red fibers (MERRF) and mitochondrial myopathy, lactic acidosis, and stroke-like episodes (MELAS)—or secondary to retinal degeneration, for example, Kearns–Sayre syndrome. Wolfram’s syndrome (see later in the chapter) is also probably the result of a mitochondrial disorder.
Autosomal dominant (juvenile) optic atrophy generally has an insidious onset in childhood, with slow progression of visual loss throughout life. It is often detected as mild reduction in visual acuity by childhood vision screening programs. There is characteristically a centrocecal scotoma with impaired color vision. Temporal optic disk pallor is usually present, although often mild, and mild disk cupping is occasionally seen. Diagnosis is by identification of other affected family members. The genetic defect has been mapped to the long arm of chromosome 3, but a diagnostic genetic test is not yet available. Rarely, the disease is associated with congenital or progressive deafness or ataxia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


