19 NEURO-OPHTHALMIC DISEASE
Pupil Abnormalities
Leonid Skorin Jr and Bruce G. Muchnick
ICD-9: 379.40
 THE DISEASE
THE DISEASE
Pathophysiology
Pupil size reflects the neural input of both the sympathetic and parasympathetic systems on the dilator and sphincter muscles, respectively, of the iris. Normal pupils are 3 to 4 mm in diameter in ambient light and are relatively equal in size. This equality in pupil size is a result of the bilateral symmetrical afferent input to the Edinger-Westphal complex in the midbrain and because of the symmetrical output of the paired sympathetic nuclei in the hypothalamus. Any disruption of the neural pathway or iris architecture will lead to abnormalities of the pupillary state.
Etiology
A relative afferent pupillary deficit (RAPD) or Marcus Gunn pupil will result from a significant unilateral or asymmetric visual deficit caused by retinal or optic nerve disease (optic neuropathy).
Anisocoria is the state where the two pupils are unequal in size. The etiology depends on whether the abnormal pupil is the dilated or constricted pupil.
Dilated pupils can occur from iris sphincter trauma, Adie’ tonic pupil, third-nerve palsy, or pharmacologic blockade. Constricted pupils can occur with iritis, pharmacologic blockade, Horner’ syndrome, Argyll Robertson pupil, or long-standing Adie’ pupil. Physiologic anisocoria is present when the pupil size difference remains the same in both bright light and in darkness.
The Patient
Symptoms and signs will vary according to the underlying etiology.
Clinical Symptoms
Patients with a RAPD will complain of vision and color loss in the affected eye.
Patients with anisocoria are often asymptomatic or may complain of other symptoms, such as droopy eyelid, blurred vision, focusing problems, or diplopia, depending on any accompanying symptoms.
Clinical Signs
Prominent Signs
- With RAPD, there will be a positive swinging flashlight test (Fig. 19-1).
- Anisocoria—Pupillary size difference that may vary between light and dark.
- Adie’ tonic pupil—The affected pupil is larger and reacts poorly to light or near stimuli.
- Argyll Robertson pupil—The miotic pupils are irregular and do not respond to light with poor dilation in the dark yet good near response.
- Pharmacologic pupil—Either miotic or dilated pupil that does not respond to any pharmacologic testing.
- Horner’ syndrome—Miotic pupil accompanied by ptosis and possibly anhidrosis. Anhidrosis will be present if the lesion involves the sympathetic pathway proximal to the bifurcation of the common carotid artery.
- Third-nerve palsy—Dilated pupil accompanied by ptosis and extraocular muscle palsies.
- In light-near dissociation (LND) of pupils, there is better pupillary response to near stimuli than to light stimuli. This is found in optic neuropathy, retinopathy, Adie’ tonic pupil, Argyll Robertson pupil, Parinaud’ dorsal midbrain syndrome, aberrant regeneration of the third nerve, and diabetes.
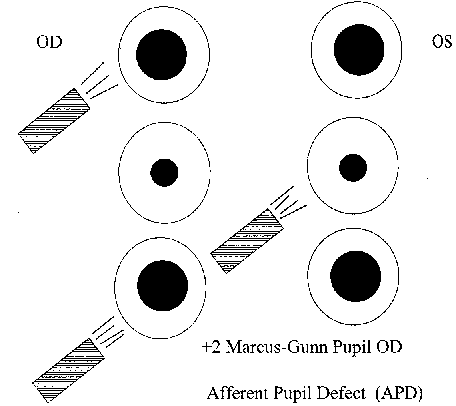
Figure 19-1. Swinging flash-light test (examiner facing patient). Right eye affected; left eye normal.
Illustration courtesy of Kathleen A. Skorin, C.O.A.
Demographics
Physiologic anisocoria is usually less than 1.0 mm and can change from day to day.
It is as common in men as in women, in the morning as in the afternoon, in the young as in the aged. The prevalence of physiologic anisocoria is approximately 20%.
Significant History
- History of optic nerve disease or retinopathy
- Recent changes in pupil size
Ancillary Tests
Swinging flashlight test: In this test, light is directed first into one pupil, then moved over quickly to the other pupil,t and repeated several times while both pupils’ response is observed. The test is done in a dimly lit room, with the patient gazing at a distant target and not at the light source. Allow just 3 to 5 seconds of illumination on each side.
When both afferent pathways are normal, the pupils remain essentially the same size no matter which side is illuminated. When an afferent defect is present, the pupils become larger when the light is directed to the involved side (see Fig. 19-1).
The RAPD can be measured subjectively or objectively with neutral density filters.
To measure an RAPD objectively, a neutral density filter is held in front of the uninvolved eye as the swinging flashlight test is performed. The filter density is increased until the pupil reactions on the swing test are equal.
Anisocoria is evaluated by determining whether pupillary inequality is caused by a defect in sympathetic or parasympathetic innervation, or is because of a structural disorder of the iris. Therefore, a comparison of pupil size by measurement should be done in bright and dim ambient illuminations (Fig. 19-2).
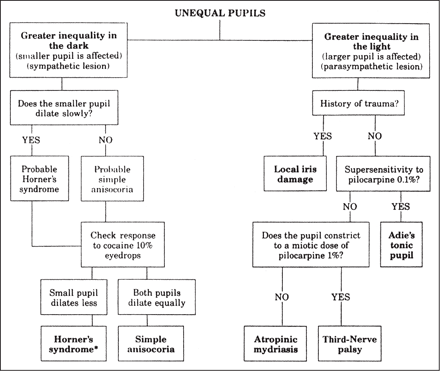
Figure 19-2. Flow chart for evaluation of anisocoria.
(Reprinted from Multack RF, Lannin WC, Olbum JR. Improving diagnostic acumen in pupillary evaluation: a review for the primary care physician. J Am Osteopath Assoc 1989;89:917–924, with permission.)
The Treatment
The treatment of pupillary abnormalities is directed to the underlying cause. (See specific sections on optic neuropathies, retinopathy, Horner’ syndrome, Adie’ tonic pupil, Argyll Robertson pupil, third-nerve palsy).
Adie’ Tonic Pupil
Leonid Skorin Jr and Bruce G. Muchnick
ICD-9: 379.46
 THE DISEASE
THE DISEASE
Pathophysiology
Tonic pupils occur from damage to the ciliary or the postganglionic short ciliary nerves that innervate the pupillary sphincter and ciliary muscles.
Selective damage causes areas of the iris to contract, with adjacent areas experiencing sector paralysis.
Etiology
Most cases of tonic pupil are idiopathic, although some follow a mild upper respiratory infection or other viral illness. Ocular and orbital trauma or surgery can also cause a tonic pupil.
If the tonic pupil is accompanied by diminished deep tendon reflexes, it is known as Holmes-Adie’ syndrome. This can be explained by concurrent involvement of the ciliary ganglion (tonic pupil) and dorsal root ganglion (areflexia).
Other causes of tonic pupil, especially if bilateral, include orthostatic hypotension, diabetes mellitus, Guillain-Barré’ syndrome, dysautonomias (such as Riley-Day’ syndrome), and acute ophthalmologic polyneuritis (Fisher’ syndrome). Other causes include infections, uveitis, sarcoidosis, orbital and intraorbital surgery, laser therapy, and trauma.
The Patient
The patient is often asymptomatic or will have accommodative difficulties with anisocoria.
Clinical Symptoms
The patient with tonic pupil may complain of pupil size difference, photophobia, or blurred near vision with difficulty focusing.
Clinical Signs
Prominent Signs
- Unilateral irregularly dilated pupil with poor to no response to light and shows a slow constriction to near stimulus with slow redilation
Subtle Signs
- Sectoral iris paralysis
- Irregular (vermiform) movements of the iris sphincter
- Defective accommodation
Demographics
Up to 80% of tonic pupils are unilateral initially, and after 1 or 2 months, a tonic pupil may become miotic and smaller than the fellow pupil. Of the unilateral cases, approximately 4% become bilateral each year.
Tonic pupils occur in younger individuals (20 to 40 years of age), and there is a female predilection, with up to 70% of cases occurring in young women.
From 50% to 90% of patients with tonic pupil will also have diminished deep tendon reflexes.
Significant History
- Recent viral illness
- Ocular or orbital trauma
- Hypotension
- Diabetes
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing, accommodation testing, slit-lamp evaluation of the iris, and fundus evaluation.
Pupil testing should be performed for light and near. To test for cholinergic supersensitivity, have the patient look at a distant target and administer pilocarpine 0.125% in each eye. Remeasure the pupil size after 15 minutes, making sure the patient does not do any near work.
The tonic pupil will constrict significantly as compared with the normal pupil.
Occasionally, an acute tonic pupil will not show cholinergic supersensitivity until several months later. No neuroimaging studies are necessary once the Adie’ tonic pupil is pharmacologically confirmed. All patients with bilateral tonic pupils, whether dilated or miotic, should have serological testing for syphilis FTA-ABS. Neurosyphilis may cause bilateral tonic pupil confirmed on cholinergic supersensitivity testing.
The Treatment
No treatment is necessary in an asymptomatic patient. If the patient is photophobic, dilute pilocarpine or tinted lenses can be helpful. Reading difficulties can be corrected with appropriate refractive correction for near work, often requiring unequal near adds. Consider colored contact lenses to hide any anisocoria.
Leonid Skorin Jr and Bruce G. Muchnick
ICD-9: 379.45
 THE DISEASE
THE DISEASE
Pathophysiology
The lesion responsible for pupillary abnormality is uncertain but may originate in the region of the Sylvian aqueduct in the rostral midbrain interfering with the light reflex fibers and supranuclear inhibitory fibers as they approach the Edinger-Westphal nuclei. The more ventrally located fibers for near response are not affected. In addition, the miosis found in classic Argyll Robertson’ syndrome can be explained by a lesion rostral to the oculomotor nuclei for pupillary constriction. This would interrupt the supranuclear inhibitory pathways from the higher brain centers thus producing a spastic paralysis in the oculomotor nuclei yielding a miotic pupil.
Etiology
The Argyll Robertson pupil has been recognized as the hallmark of late central nervous system (CNS) syphilis. Other possible causes include tabes diabetica, multiple sclerosis, encephalitis, sarcoidosis, chronic alcoholism, trauma, neoplasm, and diabetes mellitus.
The Patient
The patient will present with abnormal pupils characterized by miosis, unresponsiveness to light stimulus, and contraction on near effort in eyes with intact visual function.
Clinical Symptoms
The patient with Argyll Robertson pupils is often asymptomatic. The condition is typically bilateral and may be either symmetric or asymmetric. With few exceptions, it is permanent and usually stable but may deteriorate toward total pupil immobility. It develops over months to years.
If because of neurosyphilis, any signs that are present may include mixed neurologic features. These include meningeal syphilis (headache, nausea, vomiting, neck stiffness), microvascular syphilis (stroke), and parenchymal syphilis (paresthesias).
Clinical Signs
Prominent Signs
- Miotic, irregular pupils that do not react to light but react briskly to a near stimulus
Subtle Signs
- Pupils dilate poorly in the dark
- Pupils dilate poorly to mydriatic agents
- Usually bilateral but asymmetric
- Iris atrophy
Demographics
Up to 80% of patients with neurosyphilis will have Argyll Robertson pupils.
Significant History
- Tertiary syphilis
- Diabetes mellitus
- Demyelinating disease
- Trauma
- Neoplasm
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing, testing pupillary reaction to light and near, slit-lamp evaluation for iris atrophy or interstitial keratitis, and fundus evaluation. Argyll Robertson pupils dilate poorly if the iris atrophy is extensive. The less the atrophy, the better the dilation. Likewise, miotics work poorly in the highly atrophied iris and well in the uninvolved iris. Iris atrophy may be visualized by transillumination of the globe.
Laboratory studies: VDRL, RPR, FTA-ABS, MHA-TP.
Treatment is indicated if there is active disease, and the patient should have an internal medicine evaluation.
Cat Scratch Disease
Joseph W. Sowka and Leonid Skorin Jr
ICD-9: 078.3
 THE DISEASE
THE DISEASE
Pathophysiology
Cat scratch disease (CSD) is an infection caused by a fastidious Gram-negative bacillus from exposure to an infected cat or kitten. The most common manifestation of CSD in humans is regional lymphadenitis. Other systemic manifestations include Parinaud’ oculoglandular syndrome (conjunctivitis, retrotarsal conjunctival granulations, regional preauricular and cervical lymphadenitis, and fever), hepatosplenic infection, encephalopathy, osteomyelitis, and endocarditis. Fever, chills, abdominal pain, malaise and backache commonly occur. In immunocompetent individuals, the disease is typically self-limiting over the course of 3 to 6 weeks.
Beyond Parinaud’ oculoglandular syndrome, the ocular manifestation most associated with CSD is neuroretinitis, a combination of disc edema with stellate macular star of exudates. CSD appears to be the most common cause of neuroretinitis. In cases of neuroretinitis, vision is often affected, but to a variable degree. Vision may range from 20/20 to count fingers. Visual field defects are typically central and cecocentral scotomas. Dyschromatopsia and a relative afferent pupil defect may be present to variable degrees. Other posterior segment findings include peripapillary serous macular detachment, discrete foci of retinitis manifested as white retinal or choroidal lesions, vitritis, and submacular exudates. Anterior uveitis may also occur.
Etiology
The causative organism of CSD is the Gram-negative rod, Bartonella henselae. However, other strains of the organism, notably Bartonella quintana, have been implicated in the disease as well. The disease is transmitted by the scratch or bite of an infected cat or kitten, which are natural hosts for these organisms. About 10% of pet cats and 33% of feral cats are bacteremic with these organisms. Kittens are more likely than adult cats to carry the blood-borne bacteria and are the more common vector. Arthropod vectors exist as well, but transmission through fleas is questionable. Fleas are, however, the most likely transmission source between cats. The likely means of transmission of B. henselae from cats to humans may be inoculation with bacteria-containing flea feces through a contaminated cat scratch wound or across a mucosal surface. There may or may not be an identified history of a scratch or bite, but contact with felines is present. Ticks can also transmit this disease at the same time as Lyme’ disease. However, CSD is often overlooked in these cases of coinfection due to similar manifestations.
After an incubation period of several days to weeks, the patient develops a self-limiting regional lymphadenopathy with a small cutaneous lesion at the site of inoculation. The patient will demonstrate fever and flu-like symptoms, which will typically resolve over 3 to 6 weeks.
The Patient
The patient is typically younger and has had exposure to cats or kittens. Often there will be a history of a bite or scratch, but this history is not always present as transmission across a mucosal surface is possible. The patient will manifest a flu-like illness with lymphadenopathy and fever.
Clinical Symptoms
- Regional lymphadenopathy
- Fever
- Malaise
- Small cutaneous lesion at site of inoculation
- Parinaud’ oculoglandular syndrome
- Painless vision loss (typically unilateral)
- May be asymptomatic
- May diminish to count fingers
- May be asymptomatic
- Dyschromatopsia
Clinical Signs
See Figure 19-3.
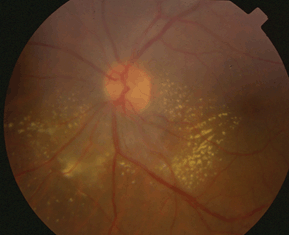
Figure 19-3. Cat scratch disease.
- Conjunctivitis
- Retrotarsal conjunctival granulations
- Regional preauricular and cervical lymphadenitis
- Stellate neuroretinitis (disc edema with macular star of exudates): Macular star may develop late in the disease
- Vision ranging from 20/20 to count fingers
- Relative afferent pupil defect (±)
- Anterior uveitis
- Focal retinochoroiditis
- Submacular exudates with overlying serous retinal detachment
- Disc edema
- Disc edema with associated peripapillary or macular serous retinal detachment
Demographics
CSD typically afflicts young, otherwise healthy adults. It occurs commonly in children. Higher incidence is reported in fall and winter, attributed to seasonal breeding of cats.
Significant History
Despite the name, a scratch or bite from a cat is not always present. However, contact or exposure to cats or kittens is invariably reported. Patients may not remember a scratch or bite, but the disease is suspected in patients developing painless vision loss or disc edema following a febrile illness.
Ancillary Tests
The definitive test for CSD is the ELISA B. henselae titer. In that there are other species that can cause CSD, it is appropriate to obtain titers for B. quintana (and possibly Bartonella clarridgeiae) as well. Sera from 95% of patients with clinically defined CSD show IgG titers of 1:64 and above. Early in the disease process, IgG may be absent, but IgM may be positive. In some cases, there will be a false-negative ELISA despite clinical signs and symptoms of CSD. An alternate diagnostic modality is a polymerase chain reaction analysis of lymphadenopathy aspirate and should be considered in the clinical situation where CSD is strongly suspected and ELISA titers are either negative or borderline.
The Treatment
In immunocompetent individuals, CSD is generally a benign self-limiting disease with an excellent prognosis with little chance for morbidity. Without ocular involvement and visual loss, medical treatment is generally unnecessary. The organism is quite susceptible to a number of antibiotics, such as penicillin, cephalosporins, aminoglycosides, tetracyclines, macrolides, fluoroquinolones, sulfamethoxazole, and trimethoprim, and rifampicin. Though antibiotics are frequently used for CSD neuroretinitis, there are no controlled clinical trials that indicate a better clinical outcome from this therapy. While the benefit of therapy in immunocompetent patients is unknown, cases complicated by ocular involvement are recommended for therapy. Azithromycin is the drug of choice if treatment is initiated. For pediatric patients, an initial dose of 10 mg/kg on the first day, followed by 5 g/kg/d for the following 4 days is the recommended regimen. For older children and adults, the recommended regimen is 500 mg on the first treatment day, followed by 250 mg/d for the next 4 days. This is most often used when there is vision loss from neuroretinitis. Whether oral prednisone has a role in the management of this disease is unknown.
Long-term prognosis for patients with ocular involvement is good, but some individuals may acquire a mild postinfectious optic neuropathy.
Horner’ Syndrome
Alan G. Kabat and Leonid Skorin Jr
ICD-9: 337.9—UNSPECIFIED DISORDER OF AUTONOMIC NERVOUS SYSTEM
ICD-9: 954.0—INJURY TO CERVICAL SYMPATHETIC NERVES
 THE DISEASE
THE DISEASE
Pathophysiology
Partial or complete interruption of the oculosympathetic pathway results in Horner’ syndrome. The oculosympathetic pathway is composed of three neurons:
- The first-order neuron arises from the posterior hypothalamus, traveling through the brainstem and cervical cord, and synapsing at the ciliospinal center of Budge at C8-T2.
- Preganglionic second-order neuron fibers exit the spinal cord with the first ventral thoracic root and continue through the stellate ganglion at the pulmonary apex to the superior cervical ganglion at the bifurcation of the internal and external carotid arteries (C3-4).
- The postganglionic third-order neuron originates from the superior cervical ganglion and travels along the internal carotid artery, penetrating the base of the skull and continuing through the cavernous sinus. The fibers that will innervate the eye and Müller’ muscles of the eyelids then enter the orbit via the superior orbital fissure. Fibers destined for the dilator muscle enter the eye via the long posterior ciliary nerves.
Etiology
- First-order neuron lesions (50% to 60%)—Cerebrovascular disease (Wallenberg’ lateral medullary plate infarction syndrome), cervical trauma, Arnold-Chiari malformation, demyelinating disease (e.g., multiple sclerosis), syringomyelia, tumors of the pons, third ventricle, cervical cord or pituitary, osteoarthritis of the neck with bony spurs.
- Second-order neuron lesions (20% to 30%)—Tumors of the chest apex (Pancoast’ tumor) or superior mediastinum, lymphadenopathy, aortic aneurysm, thyroid enlargement, surgical intervention (thyroidectomy, radical neck surgery, carotid angiography).
- Third-order neuron lesions (20%)—Vascular headache syndromes (migraine, cluster, Raeder’ paratrigeminal neuralgia), cavernous sinus or middle cranial fossa lesions, basal skull fracture, carotid-cavernous fistula, nasopharyngeal carcinoma, otitis media, sinusitis, trigeminal herpes zoster, Tolosa-Hunt’ syndrome, carotid artery occlusion or dissection.
- Congenital—Birth trauma sustained during labor or delivery, cervical and mediastinal neuroblastoma.
The Patient
The classic Horner’ syndrome triad consists of unilateral ptosis, miosis, and anhidrosis (Fig. 19-4).
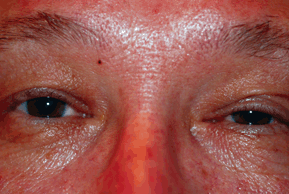
Figure 19-4. Horner’s syndrome. Ptosis, miosis on left side.
Clinical Symptoms
A patient with a recent onset of Horner’ syndrome will complain of a droopy eyelid and a lack of sweating on one side of the face, provided the lesion occurs distal to the carotid bifurcation, since most of the sudomotor sympathetic fibers to the face travel with the external carotid artery.
Clinical Signs
Prominent Signs
- Anisocoria, which is greater in dim illumination
- Upper eyelid ptosis
- Diminished iris pigment in the affected eye (heterochromia iridis), if congenital
Subtle Signs
- Upside-down ptosis or elevation of the lower eyelid
- Apparent enophthalmos from a narrowed palpebral fissure
- Dilated conjunctival and facial vessels
- Decreased intraocular pressure
- Increased accommodative amplitude
- Absence of dilation to psychosensory stimuli
Demographics
Horner’ syndrome can occur in any patient regardless of gender or age. In those 20 years and younger, trauma is the most significant cause. From 21 to 50 years, almost half the cases result from tumors. In patients over 50 years of age, neoplasia is the most important cause. Horner’ syndrome is the initial sign of malignancy in 35% of patients. Overall, the cause of Horner’ syndrome can be identified in only about 60% of patients.
Significant History
- New onset of headache
- Trauma
- Recent cardiac, neck, or thoracic surgery
- Recent stroke
Ancillary Tests
Ocular assessment includes visual acuity, extraocular motility, levator function, pupil testing, visual fields, biomicroscopy, and fundus evaluation. Examination of old photographs may be helpful in determining the duration of the condition. Definitive diagnosis is based on pharmacologic pupillary testing to cocaine and hydroxyamphetamine.
- Cocaine test. Cocaine produces dilation of the pupil by preventing reuptake of norepinephrine. If the sympathetic innervation to the eye is interrupted at any level (central, preganglionic, or postganglionic), cocaine will have no dilation effect. Cocaine helps confirm the presence or absence of oculosympathetic palsy by increasing pupillary inequality; however, this test does NOT indicate the location of the lesion. (Note: patients may test positive on drug testing for 48 hours following the cocaine test.)
- Cocaine (4% to 10%), one drop is placed in each eye and then repeated in several minutes. The pupils should be evaluated after 50 to 60 minutes. A Horner’ pupil does not dilate as well as the normal pupil.
- Hydroxyamphetamine test. Hydroxyamphetamine (Paredrine) 1% causes a release of norepinephrine from the nerve endings at the myoneural junction, stimulating the dilator muscle. This helps distinguish a third-order neuron from a first-order neuron and second-order neuron. Third-order neuron lesions are considered to be “benign” while first-and second-order lesions are potentially life threatening.
- Hydroxyamphetamine testing should not be performed for 48 hours following the cocaine test.
- Hydroxyamphetamine 1% is placed in each eye, repeating one drop 1 minute later. The pupils are evaluated in 30 minutes. The normal pupil will dilate, while a third-order neuron lesion pupil will show no dilation. Pupillary dilation will be normal if the Horner’ syndrome is a result of a lesion to the first-order or second-order neuron.
- Laboratory studies. Lab studies are not diagnostic of Horner’ syndrome, but may aid in identifying the underlying etiology. Complete blood count (CBC) with differential, syphilis testing (FTA-ABS and RPR), and purified protein derivative for tuberculosis should be considered. Imaging studies may include chest x-ray with lordotic view to image the lung apex, computed tomography (CT) scan, or magnetic resonance imaging (MRI) of the brain, and x-rays of the cervical spine.
If a central lesion is suspected, the patient should be referred for a neurology consult. Likewise, if malignancy is suspected, referral to an internist or oncologist is indicated. Vascular lesions warrant consultation with a vascular or thoracic surgeon.
The Treatment
Postganglionic lesions usually do not need treatment. Central or preganglionic lesions need to be addressed with appropriate consultation. Neurosurgical intervention may be required for potentially life-threatening situations. Vascular headache syndromes (e.g., migraine, cluster) should be treated to minimize recurrence.
Oculomotor Nerve Palsy
Joseph W. Sowka and Leonid Skorin Jr
ICD-9: 378.52
 THE DISEASE
THE DISEASE
Pathophysiology
The oculomotor (third) cranial nerve innervates five extraocular muscles (the levator, superior, inferior and medial recti, and the inferior oblique) and carries the parasympathetic outflow to the ciliary ganglion, controlling pupillary constriction and accommodation. Any mechanism that disrupts this outflow will cause partial or complete oculomotor nerve palsy. Additionally, pupil dysfunction may also ensue.
Etiology
Causes of nuclear oculomotor nerve palsy include multiple sclerosis, vascular accidents, tumors, and infections. Midbrain vascular accidents account for the majority of fascicular palsies, involving both the oculomotor nucleus as well as the nerve’ fascicles as they course through the parenchyma of the midbrain. After exiting the brainstem in the interpeduncular fossa, the oculomotor nerve passes inferolateral to the posterior communicating artery (PCA) and medial to the edge of the tentorium. In this region, it is vulnerable to damage from transtentorial herniation also known as uncal syndrome. Compression from an aneurysm of the PCA can also cause pupil-involving oculomotor palsy. Nerve damage can also result from subarachnoid hemorrhage and meningitis.
The disease processes within the cavernous sinus usually involve more than one nerve and include neoplasm, aneurysm, thrombosis, fistula, inflammation, and ischemia.
The oculomotor nerve divides into superior and inferior divisions at the level of the anterior cavernous sinus or superior orbital fissure. Superior-division palsies, involving the superior rectus and levator, may result from aneurysm, viral infection, diabetes, and enlargement of the third ventricle. Inferior-division palsies, involving the inferior rectus, inferior oblique, medial rectus, and ciliary sphincter, result from trauma, viral infections, local orbital disease, and brainstem vascular malformations. Orbital involvement, such as trauma, tumor, aneurysm, and infection from a tooth abscess, can damage the oculomotor nerve.
Aberrant regeneration or oculomotor synkinesis occurs only with mechanical disruption of fibers following an acute palsy such as from trauma or aneurysmal rupture and never from vasculopathic palsies. This type of misdirection can also be found in congenital oculomotor nerve palsy and ophthalmoplegic migraine. Patients with ophthalmoplegic migraine are children and young adults who have a strong family history of migraine.
The Patient
Symptoms and signs depend on the location of the lesion that is disrupting the oculomotor nerve. Identifying pupil involvement is critical to diagnosis and management.
Clinical Symptoms
The patient with oculomotor nerve palsy will complain of diplopia, droopy eyelid, difficulty focusing or reading, and possible eye or hemicranial pain (Fig. 19-5).
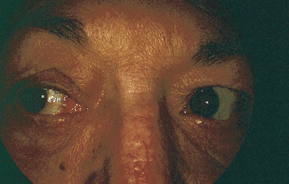
Figure 19-5. Oculomotor nerve palsy. Adduction deficit in partial oculomotor palsy.
Clinical Signs
Prominent Signs
- Complete palsy, which includes ptosis and restriction of ocular movement except for abduction and intorsion. The pupil may also be fixed and dilated.
- Superior-division palsy includes ptosis with a restriction in upgaze.
- Inferior-division palsy includes restriction in downgaze, restricted adduction, and pupil dilation.
- Nuclear-fascicular palsies may also involve contralateral hemiplegia, intention tremor, and ataxic gait depending upon the location of the damage within the midbrain.
Subtle Signs
- Aberrant regeneration or oculomotor synkinesis
- Loss of accommodation
- Loss of near-light reflex
Demographics
Approximately 20% of oculomotor third-nerve palsies are a result of expansion or hemorrhage of an aneurysm, and vasculopathic causes, such as diabetes, account for another 20%. Patients who have an underlying vasculopathic etiology are usually older than 45 years of age, and 60% or more are diabetic. Only 15% of oculomotor palsies are because of neoplasm. In children, 40% of oculomotor palsies are congenital, and 20% are because of trauma.
The pupil is spared in 80% of vasculopathic oculomotor nerve palsies and involved in 95% of aneurysmal palsies. Patients developing oculomotor nerve palsy from aneurysmal compression may initially not present with anisocoria or pupil involvement but may evolve and develop pupil dilation over several days. Lesions in the cavernous sinus also may spare the pupil or present with a concurrent Horner’ syndrome. Approximately one third of patients with CN III palsy from vascular infarct manifest a small degree of anisocoria of typically less than 1 mm. In contradistinction, aneurysmal compression typically causes more than 2 mm of anisocoria.
Significant History
- Diabetes
- Hypertension
- Hyperlipidemia
- Herpes zoster
- Leukemia
- New onset headache
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing, visual field, exophthalmometry, levator function, and pupil assessment.
When the pupil is not involved, an edrophonium test, anticholinesterase antibody assay, evaluation of orbicularis strength, and evaluation of ptosis after sustained upgaze should be performed to rule out myasthenia gravis (MG) mimicking oculomotor palsy.
Neuroimaging with a CT or MRI and magnetic resonance angiography (MRA), lumbar puncture, or cerebral angiography is indicated for all pupil-involved oculomotor palsies or pupil-sparing palsies if the patient is less than 45 years old with no vasculopathic history, has incomplete palsy, has a palsy that has not shown any improvement over 3 months time, or has more than just an isolated oculomotor deficit. Any patient who develops aberrant regeneration should also have neuroimaging.
Laboratory studies: CBC with differential, glucose tolerance test, erythrocyte sedimentation rate (ESR), RPR, antinuclear antibody. A blood pressure measurement should also be done.
The Treatment
Most patients with ischemic oculomotor nerve palsies show improvement within 1 to 2 months and maximally by 6 months. In ischemic vascular CN III palsy, the pupil will not evolve and aberrant regeneration will not occur. While waiting for improvement to occur, the underlying vasculopathic abnormality should be corrected, and the patient may wear a temporary eye patch for any disturbing diplopic symptoms. Patients who have pupil-involving oculomotor palsies will need emergent evaluation and testing with possible hospitalization. If an intracranial aneurysm is the suspected cause of the oculomotor palsy, then the patient should be sent emergently to the hospital, as 20% of these patients die within 48 hours because of aneurysm rupture and subsequent subarachnoid hemorrhage. Close pupil observation is required as pupil involvement may be delayed by 5 to 7 days. This is especially true for patients with incomplete oculomotor nerve palsy as they are more likely to have an insipient aneurysm developing. Complete and total oculomotor palsy in an adult over age 50 without pupil involvement is rarely ever caused by an aneurysm.
Children with chronic oculomotor palsies may need Fresnel prism correction or botulinum toxin injection to the lateral rectus to prevent contracture of the muscle. Surgery should be delayed until 6 months from the time improvement has ceased. Ipsilateral horizontal recess-resect procedures work well if there is at least 50% medial rectus function. If the palsy is complete, large horizontal recess-resect procedures are combined with contralateral lateral rectus weakening and/or ipsilateral superior oblique transposition. Contralateral vertical muscle surgery may also need to be performed.
Trochlear Nerve Palsy
Joseph W. Sowka and Leonid Skorin Jr
ICD-9: 378.53
 THE DISEASE
THE DISEASE
Pathophysiology
The trochlear (fourth) cranial nerve is the only cranial nerve that exits at the dorsal aspect of the brainstem. The nerve fibers then cross and follow a long and circuitous course from the brainstem to the superior oblique muscles. This makes the nerve vulnerable to injury at many locations.
Etiology
Causes of nuclear-fascicular trochlear nerve palsy include ischemia from infarction, hemorrhage, demyelination, compression, and trauma. Bilateral trochlear nerve palsies are often seen following blows to the head. Bilateral, and occasionally unilateral, trochlear nerve palsies can be associated with a tumor of the pineal gland compressing the posterior midbrain in the dorsal midbrain syndrome. Loss of upward saccades and a light-near dissociated pupil accompany the trochlear palsy in dorsal midbrain syndrome. Distention of the fourth ventricle or contrecoup forces transmitted to the brainstem by the free tentorial edge may injure the nerves. As the nerve courses through the cavernous sinus and orbit, it can be affected by inflammation, tumor, aneurysm, fistula, or trauma. Isolated trochlear nerve palsies may be congenital and appear in late childhood to mid-adult life as a result of decompensation. Otherwise, most isolated acquired trochlear nerve palsies are due to trauma, hypertension, atherosclerosis, and diabetes. More rare causes of palsy include herpes zoster ophthalmicus, collagen vascular disease, aneurysm, hydrocephalus, multiple sclerosis, polycythema vera, cat-scratch disease, and encephalitis.
The Patient
Most patients with trochlear nerve palsy will complain of vertical diplopia that is eliminated when one eye is closed (Fig.19-6). Often, head tilting to the opposite side will relieve the diplopia or make the patient feel more comfortable.
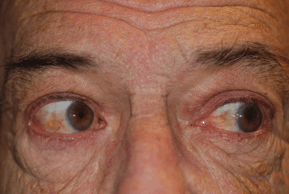
Figure 19-6. Trochlear nerve palsy. Right fourth nerve palsy. Right overaction on left gaze.
Clinical Symptoms
- Vertical diplopia that is worse in the field of action of the involved superior oblique muscle
- Difficulty with near tasks or downgaze
Prominent Signs
- Positive Parks “three-step test”
Step 1: Vertical deviation on the side of the lesion
Step 2: Vertical deviation worsens on gaze away from the involved nerve
Step 3: On Bielschowsky head-tilt testing, the deviation is greater on head tilt toward the involved trochlear nerve
Subtle Signs
- Assumed or habitual head tilt toward the contralateral shoulder
- Large vertical fusional amplitudes
- Excyclotorsion as measured by double Maddox rod test
- Hyperdeviation greater on downgaze
Demographics
The most common cause of trochlear nerve palsy is trauma, which constitutes approximately 40% of the cases. In spite of the availability of improved diagnostic techniques, the number of cases remaining without a specific diagnosis is still approximately 30%. Ischemia causes 20% and tumors and other sinister causes constitute 10% of acquired, isolated trochlear palsies.
Congenital trochlear palsies are also very common and can present in later life due to decompensation. Approximately 30% of all trochlear palsies are bilateral, and the prevalence increases to 40% in those secondary to trauma.
Significant History
- Head trauma, especially blows to the vertex
- Hypertension
- Diabetes
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing including the Parks three-step test and Bielschowsky’ head-tilt testing. Measurement for excyclotorsion can be done with a double Maddox rod in a trial frame or phoropter, Maddox wing, or Lancaster red/green test. Cyclotorsion can also be evaluated with fundus examination or photography. Normally, the optic nerve is just slightly higher than the foveal area. In trochlear nerve palsy, the optic disc is significantly higher than the fovea on fundus photography and lower on indirect ophthalmoscopy.
If congenital trochlear nerve palsy is suspected, then measurement of the amplitude of vertical vergence should be done. In most patients, the normal value of vertical fusional amplitude is from 3 to 6 prism diopters. Patients with congenital trochlear palsy may be able to fuse anywhere from 10 to 30 prism diopters of deviation (using vertical prism bars). Evaluate possible long-standing palsy by reviewing old photographs, looking for the presence of head-tilting. Further, immediately upon entering the examination room, unobtrusively look for the presence or absence of a head-tilt.
Laboratory studies: Glucose tolerance test, ESR, C-reactive protein (CRP), blood pressure measurement, edrophonium test, and/or anticholinesterase antibody assay if MG is suspected.
If the palsy has not resolved or improved in 4 months, or if any other neurologic signs are present, then the patient will also need neuroimaging, lumbar puncture, and cerebral angiography. If the patient has developed trochlear nerve palsy from recent head trauma, the patient should undergo neuroradiological studies of the head to dismiss the possibility of a concurrent subarachnoid hemorrhage. In patients under age 40 with a sudden onset of trochlear nerve palsy, no ischemic vascular diseases such as diabetes and hypertension, and there is no evidence that the diplopia is a result of decompensation of a longstanding condition, then neuroimaging is indicated.
Because many acquired trochlear nerve palsies spontaneously recover in 4 to 6 months, observation with patching for symptomatic diplopia is sufficient. If the deviation remains stable, prisms incorporated into glasses may be of benefit. Surgical correction has a high success rate. Surgery is undertaken when the deviation has been stable for at least 6 months. Botulinum injections may be considered as well.
Abducens Nerve Palsy
Joseph W. Sowka and Leonid Skorin Jr
ICD-9: 378.54
 THE DISEASE
THE DISEASE
Pathophysiology
The abducens (sixth) cranial nerve originates in the dorsal midpons with its nucleus being surrounded by the seventh-nerve fascicle. This complex anatomic relationship helps determine the presence of associated findings. The abducens nerve fascicle travels anteriorly through the pons, lateral to the parapontine reticular formation, and through the pyramidal tract to exit the brainstem into the subarachnoid space. Due to the close anatomic relationships between these structures, isolated abducens palsy due to damage within the midpons is unlikely. The subarachnoid space is the most common locus of isolated, unilateral abducens nerve palsy. The abducens nerve then continues up the clivus, over the petrous ridge along the base of the skull, entering the cavernous sinus and the orbit to innervate the lateral rectus muscle.
Etiology
Causes of nuclear-fascicular abducens nerve palsies include tumors, demyelination, and vascular disease. The abducens nerve is vulnerable in the subarachnoid space from trauma and tumors. It is here that the nerve is affected by elevated intracranial pressure by downward displacement of the brainstem and secondary stretching of the nerve. This will manifest often as a bilateral abducens palsy with concurrent papilledema. Inflammation of the petrous bone and its dura may occur secondary to middle-ear infections, known as Gradenigo’ syndrome. Traumatic abducens nerve palsies may result from basilar skull fractures. Masses invading the base of the skull from the nasopharynx or at the cerebellopontine angle may cause abducens-nerve pareses.
Cavernous sinus and orbital etiologies include trauma, diabetes, hypertension, tumor, aneurysm, fistula, inflammation, and cellulitis. Isolated abducens nerve palsy is seen in young patients with postviral syndrome and in adults with hypertension or diabetes with ischemic mononeuropathy.
The Patient
Most patients with isolated abducens nerve palsy will complain of horizontal diplopia, which is greater at distance and is eliminated when one eye is closed. There are three distinct patient groups experiencing abducens palsy: children, young adults, and older adults.
Clinical Symptoms
Horizontal diplopia, worse at distance than near and exaggerated when the patient looks in the direction of the affected lateral rectus muscle.
Clinical Signs
Prominent Signs
- Abduction deficit
- Esotropia greater with paretic eye fixing a distant target, compared with unaffected eye fixing target
- No restriction on forced duction testing
- Millard-Gubler—Ipsilateral abducens, and facial-cranial nerve palsy with contralateral hemiparesis
- Raymond-Cestan—Ipsilateral abducens palsy with contralateral hemiparesis
- Foville’—Ipsilateral gaze palsy; ipsilateral trigeminal and facial-cranial nerve palsy with ipsilateral Horner’ syndrome
- Gradenigo’—Ipsilateral trigeminal pain, abducens, facial-cranial nerve palsy, and hearing loss
- Pseudo-Gradenigo’—Ipsilateral abducens palsy, ipsilateral trigeminal pain, ipsilateral ocular irritation, and hearing loss
Demographics
Abducens nerve palsies occur more frequently than do palsies of the oculomotor or trochlear cranial nerve. Up to 30% of the time, the exact cause of abducens palsy remains unknown, while the rest of the time there appears to be an even distribution between tumor, vasculopathic causes, and trauma. Bilateral abducens nerve palsies rarely result from vascular disease and are more common in children than in adults. Bilateral abducens palsies, especially with concurrent optic disc edema, strongly suggest increased intracranial pressure.
Significant History
- Head trauma
- Diabetes
- Hypertension
- New onset of headache, tinnitus, transient visual obscurations, nausea if increased intracranial pressure is suspected
- Possible associated hemi-paresis, facial nerve paralysis, Horner’ syndrome, otitis media
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing, forced duction testing, corneal sensitivity with cotton wisp, orbicularis strength, slit-lamp exam, and funduscopic evaluation for papilledema.
Laboratory studies: CBC with differential, glucose tolerance test, ESR, CRP, RPR, antinuclear antibody, and blood pressure measurement.
Neuroimaging with MRI if a lesion of the brainstem or posterior fossa is suspected. CT scanning is preferred if sinus or mastoid disease is suspected. Lumbar puncture and cerebral angiography may also be necessary.
Edrophonium testing and/or acetylcholinesterase antibody assay is indicated if myasthenia is a concern.
In children, nearly half of all abducens palsies are due to a CNS lesion. Neurologic evaluation, neuroimaging, and consultation is urgent in this group and the cause of the palsy shouldn’ be presumed to be benign.
In younger adults, CN VI palsy is likely to be caused by CNS mass lesions and multiple sclerosis. Thus, neuroimaging is highly recommended in this group.
There is a very low risk of acute isolated abducens palsy in an older adult being cause by an aneurysm, neoplasm, or demyelization. Isolated abducens palsy in older adults with a history of diabetes or hypertension do not need neuroimaging or other extensive evaluation unless the palsy progresses, fails to improve over 3 months, or other neurologic complications develop.
The Treatment
In an acute phase of an abducens cranial nerve palsy, simple occlusion with patching or temporary prisms if the deviation is small and not greatly noncomitant may be all that is necessary. Botulinum toxin injections into the ipsilateral medial rectus muscle may be necessary if the recovery is prolonged or the deviation is too large for prism correction or to prevent secondary contracture of the medial rectus. Surgical correction can be done after measurements have been stable for at least 6 months. An ipsilateral recess-resect procedure combined with a contralateral medial rectus recession may be indicated.
Any underlying identified medical problem must be addressed.
Facial Nerve Palsy
Leonid Skorin Jr
ICD-9: 351.0
 THE DISEASE
THE DISEASE
Pathophysiology
Facial palsy is the most common of the cranial neuropathies. Bell’ palsy is a facial palsy of no obvious cause whose presumed mechanism is swelling of the peripheral portion of the facial or seventh cranial nerve because of immune or infectious disease. The nerve becomes compressed and ischemic in its narrow course through the temporal bone. This mechanical compression causes interruption of the nerve’ transmission, resulting in a conduction block and nerve degeneration. Additional immunological response may result in fragmentation and demyelination of the nerve sheath, causing loss of function. Because this condition is temporary, remyelination occurs after the inflammation resolves.
Etiology
True Bell’ palsy or idiopathic facial palsy has no known underlying cause. Recent findings now strongly point to herpes simplex virus type I as the most likely cause of Bell’ palsy. Other viral infections such as varicella-zoster, Epstein-Barr (infectious mononucleosis), measles, rubella, rabies, mumps, cytomegalovirus, infectious hepatitis, and human immunodeficiency are also known to cause facial palsy.
Herpes zoster can cause up to 13% of facial palsies. Herpes zoster infection of the geniculate ganglion with typical herpetic vesicular eruptions is known as Ramsay Hunt’ syndrome (herpes zoster oticus).
Bacterial infection is the cause of facial palsy up to 4% of the time. Tetanus, brucellosis, typhoid fever, leptospirosis, and diphtheria are rare examples. In endemic areas, up to 25% of acute facial palsies may be caused by Lyme’ disease. Lyme’ disease is a systemic spirochetal infection with Borrelia burgdorferi that follows the bite of the deer tick.
Facial palsy can be the result of middle-ear disease (otitis media with or without mastoiditis); malignancies of the parotid gland because the facial nerve runs through the gland; and neoplasms, both primary, such as schwannomas, or secondary, such as acoustic neuromas, which cause compression at the cerebellopontine angle. Up to 21% of the time, trauma is the cause. Primary neurologic disorders, such as Guillain-Barré and Melkersson’ syndrome, may also cause facial palsy.
The Patient
Symptoms and signs may range from subtle weakness and facial muscle paresis to complete unilateral paralysis.
Clinical Symptoms
The patient with Bell’ palsy complains of sudden unilateral facial weakness. There may be drooling after brushing the teeth or when drinking, an asymmetric appearance of the mouth, an inability to whistle, and excessive epiphora. The patient may describe deadness, loss of feeling, or numbness. Up to 50% of patients experience pain behind the ear, sometimes even preceding physical changes. Hyperacusis (painful response to sound) and dysgeusia (distorted taste) are also common.
Clinical Signs
Prominent Signs
- Complete unilateral facial paresis or paralysis involving the forehead, orbicularis oculi, facial muscles of expression, and mouth (Figs. 19-7 and 19-8).
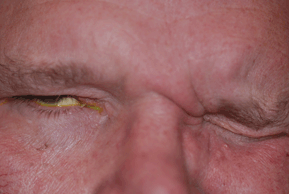
Figure 19-7. Facial nerve palsy. Bell’s palsy, right side. Inability to close right eyelid.
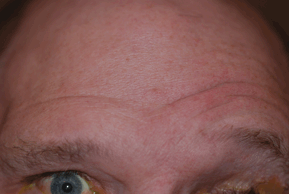
Figure 19-8. Facial nerve palsy. Bell’ palsy, right side. Inability to furrow right frontalis muscles.
Subtle Signs
- Loss of facial creases and folds
- Widening of the palpebral aperture
- Lower lid ectropion
- Inability to blink
- Down turning of one side of mouth
- Corneal irritation from dry eye and exposure
Demographics
Bell’ palsy is the most common peripheral facial palsy, occurring in 20 per 100,000 to 40 per 100,000 persons annually. Men and women of all ages are equally affected, with the majority of patients between the ages of 20 and 40. The condition affects the left and right sides of the face with equal frequency. It is bilateral at onset in about 0.3% of cases and recurrent in up to 11%. Up to 31% of patients with recurrent Bell’ palsy are diabetics. A family history can be obtained in 2% of patients. The disease is 3.3 times more common in pregnant women and is especially prevalent in the third trimester or within the first week postpartum.
Significant History
- Prior exposure to stress, upper respiratory infection, cold, fever, dental extraction, menstruation, or driving with cold air blowing on the face
- Pregnancy
- Diabetes
- Hypertension
- Exposure to tick bite and skin rash (erythema chronicum migrans)
- Medical history of Guillain-Barré, Lyme’ disease, sarcoidosis, carcinomatous, treponemal, mycobacterial or cryptococcal meningitis, multiple sclerosis, if bilateral palsy is present
Ancillary Tests
Ocular assessment includes visual acuity, extraocular muscle testing, noting any muscle imbalance or nystagmus, corneal sensitivity with cotton wisp, slit-lamp exam with fluorescein, and rose bengal staining, Shirmer basal secretion test, orbicularis strength, and Bell’ phenomenon. Otologic evaluation of the ear canal and tympanic membrane, looking behind the ear, hearing test in suspected cases of Ramsay Hunt’ syndrome.
Ancillary studies include CBC with differential, urinalysis, plasma glucose, ESR, chest x-ray, and Lyme titer. In patients with complete paralysis, assessment of neural status by electrical tests is useful. These include maximal nerve stimulation, nerve conduction, electromyography (EMG), and electroneuronography (ENOG). Imaging studies (MRI, CT) are indicated when the history or physical examination turns up suspicious findings or when facial movement does not recover within 6 months.
The Treatment
The one essential intervention in any facial palsy is corneal protection. Nonpreserved artificial tears during the day and bland ointment at night are mandatory. The patient may also physically close his or her eyelids when the eye is irritated. Moisture chambers, eyelid taping, external eyelid prosthesis, punctal plugs, botulinum toxin injection to induce upper eyelid ptosis, surgical placement of a stainless steel spring or gold weight to reanimate the upper eyelid, or partial tarsorrhaphy should be considered in cases of corneal compromise.
If the patient is seen within 2 weeks of onset of symptoms, oral prednisone 1 mg/kg of body weight daily (60 mg/d maximum) for 7 days, split into twice-daily dosing, then tapered over a subsequent 3 days can be started. Oral prednisone is most beneficial in patients with pain and complete paralysis. If seen within the first 3 days of symptom onset, oral antiviral agents (acyclovir 400 mg five times daily for 10 days, valacyclovir 500 mg three times daily for 7 days, or famciclovir 500 mg three times daily for 7 days) may be added to suspected herpes simplex palsies. The Quality Standards Subcommittee of the American Academy of Neurology recommends that the use of steroids is probably effective and under most circumstances should be considered but that antiviral agents should only possibly be considered.
Oral antiviral agents should be used in all cases of Ramsay Hunt’ syndrome. Lyme facial palsy is treated with amoxicillin, 500 mg four times a day for 3 to 4 weeks, or doxycycline, 100 mg two or three times a day for 3 to 4 weeks. Ceftriaxone, a third-generation cephalosporin, is used in amoxicillin and doxycycline failures.
Middle cranial fossa surgical decompression is of benefit in cases of complete acute paralysis between 3 and 14 days after onset, ENOG with more than 90% degeneration, and voluntary EMG with no motor-unit potentials.
Cerebral Venous Thrombosis
Bruce G. Muchnick
ICD-9: 325
 THE DISEASE
THE DISEASE
Pathophysiology
Cerebral venous thrombosis (CVT) describes the presence of a blood clot, or thrombus, within the venous system of the brain. This may lead to partial or complete occlusion of a cerebral vein, resulting in neurologic and visual dysfunction.
The blocking of a cerebral vein or dural sinus causes blood to be forced retrograde into the capillaries. This blocks oxygenated blood to the cerebral tissues, leading to ischemia, edema, and hemorrhagic infarction.
Etiology
A thrombus is an insoluble mass formed in the vascular system and composed of constituents of the blood. It forms on and is attached to the endothelium of the blood vessel. It may partially or completely occlude the lumen of the vessel.
The thrombus is composed primarily of platelets and fibrin. There may be an inflammatory response in the vessel wall accompanying the blood clot.
CVT is caused by infectious or noninfectious processes. Acute inflammation can reach the cerebral veins and arise from infections of the mastoid air cells and paranasal sinuses. Infectious sources include fungi, virus (HIV), parasites (trichinosis), and bacteria (syphilis).
Aseptic occlusion of the intracerebral venous structures can be caused by trauma, tumors, systemic diseases (lupus and Behçet’ disease), hypoperfusion (heart failure), and most commonly hypercoagulability.
No matter what the cause, cerebral venous occlusion results in hemorrhagic infarction of brain tissue and cerebral and subarachnoid hemorrhage.
Cerebral sinovenous thrombosis may be caused by closed head injury, with the stroke occurring usually within 4 days of the injury. Other causes include the use of oral contraceptives and pregnancy, but in 30% of cases, no etiology is discovered.
Dietary fasting has been shown to increase the frequency of CVT. Other risk factors include the human immunodeficiency virus (HIV), developmental venous anomalies, inflammatory bowel disease, and inner ear infection.
The Patient
Clinical Symptoms
A low grade venous sinus thrombosis may be so mild as to produce only an increase in intracranial venous pressure, resulting in a mild headache. However, the most common presenting complaint in adults is severe headache. Focal seizures may occur if there is partial ischemia to the cerebral cortex. These seizures are sometimes preceded by a prodrome of numbness, weakness, behavioral changes, and headache. A variety of visual field defects, either permanent or transient, may result from venous sinus thrombosis.
Clinical Signs
Depending on the location of the thrombosis, a wide range of neurological deficits may accompany cerebral venous thrombosis, including aphasia, memory loss, chills, sweats, and tachycardia. Ocular manifestations include pain around the eyes, orbital congestion, lid swelling, proptosis, and ophthalmoplegia.
An increase in cerebral spinal fluid level may result in papilledema.
The most common presenting signs and symptoms in children with CVT include headache, vomiting, visual disturbance, papilledema, fever, and sixth-nerve palsy. Half of all children (age 1 month to 17 years) had adverse outcomes following treatment, including chronic intracranial hypertension, residual hemiparesis, permanent sixth-nerve palsy, and death.
Cerebral venous thrombosis is more common in women, who develop the condition younger than men and more commonly have the symptom of headache upon presentation. One possible explanation is that 65% of women have a gender-specific risk factor, such as oral contraceptives, pregnancy, or hormonal replacement.
Demographics
Overall, CVT occurs in three to four cases per million people per year worldwide; in children, the incidence doubles to seven cases per million. Because of the introduction of antibiotics, septic thrombosis has become relatively rare and accounts for about 10% of all cases of cerebral venous thrombosis in the Western world. In developing countries, the rate is higher (25%). The most common cause of aseptic venous thrombosis is hypercoagulability, most commonly from the use of oral contraceptives. One study showed that in cases of cerebral venous thrombosis, 12% were deceased within 3 years, 12% had seizures, 10% had motor deficits, 8% had visual field defects, 45% had headaches, and 83% were living independent lifestyles.
- Inquire about the sudden onset of neurological deficits.
- Is the patient taking medications that may contribute to hypercoagulability states (birth control pills)?
- Is there any genetic disorder in the family or patient that may lead to a hypercoagulability state?
- Has there been any blunt injury to the head?
- Did the patient receive electroconvulsive treatment?
- Does the patient have any known systemic disorders?
- Does the patient have any signs of infection of the head or neck?
Ancillary Tests
The primary method of diagnosing cerebral venous thrombosis is noninvasive neuroimaging. This includes CT scanning and MRI. CT scanning may be performed unenhanced or with injection of iodinated contrast material. T1-and T2-weighted MR images in patients with venous thrombosis reveal abnormalities in the veins and sinuses.
MR angiography (3D MR flow imaging) can reveal abnormalities in the intracranial and extracranial vasculature. Conventional (invasive) angiography can then confirm the diagnosis of thrombosis.
Laboratory tests (CBC with differential, blood cultures) should be utilized to rule out sepsis.
Blood tests are helpful to detect a hypercoagulability state. These include CBC, ESR, CRP, prothrombin time, partial thromboplastin time, protein S deficiency, protein C deficiency, antithrombin III, factor V Leiden (activated protein C resistance), anticardiolipin profile, lupus anticoagulant, and homocysteine.
The Treatment
Treatment depends on the underlying cause of the thrombosis and location and severity of the neurological deficit.
- Septic thrombosis requires antimicrobial treatment and possible surgery
- Blood thinners (Heparin) are used in many cases to treat aseptic thrombosis
- Urokinase (thrombolysis) can be used in aseptic thrombosis
- Surgical removal of the thrombus: Decompressive hemicraniectomy improves morbidity and mortality in severe CVT
- Stent placement
- Treatment of elevated ICP (acetazolamide), lumbar punctures, or shunt procedures
- Long-term blood-thinner therapy (Warfarin)
- Men have an average of 71% complete recovery
- Women have a better prognosis than men (81% exhibit complete recovery).
- After 5 years, patients report long-term problems including impaired concentration (75%), headache (43%), depression (30%), and fatigue (30%); and 20% were unemployable.
- Thirteen percent of patients have a poor clinical outcome.
Cavernous Sinus Syndrome
Leonid Skorin Jr and Bruce G. Muchnick
ICD-9: 437.6—NONPYOGENIC THROMBOSIS OF INTRACRANIAL VENOUS SINUS
ICD-9: 325—PHLEBITIS AND THROMBOPHLEBITIS OF INTRACRANIAL VENOUS SINUS
 THE DISEASE
THE DISEASE
Pathophysiology
The cavernous sinuses are triangular interconnecting structures, the walls of each sinus extending from the superior orbital fissure to the posterior wall of the sella and created by dura mater. Each sinus contains the oculomotor, trochlear, abducens, and trigeminal cranial nerves. Other important structures include the sympathetic carotid plexus and the intracavernous carotid artery.
Etiology
The causes of cavernous sinus syndrome are varied. They include trauma; carotid-cavernous fistula; dural-cavernous fistula; pituitary adenoma or other neoplasm, either primary or metastatic, such as lymphoma or leukemia; inflammation from bacterial cause, such as sinusitis, viral, such as herpes zoster, fungal, such as mucormycosis, or idiopathic, such as Tolosa-Hunt’ syndrome.
The Patient
Symptoms and signs are those of multiple cranial nerve palsies with pain and possible pupil involvement.
Clinical Symptoms
The patient may complain of double vision, pain that may be referred to the orbit or supraorbital region, photophobia, anisocoria, and eyelid drooping.
Clinical Signs
Prominent Signs
- Painful ophthalmoplegia
- Dilated pupil
- Horner’ syndrome
- Exophthalmos
- Conjunctival chemosis
- Dilated episcleral and conjunctival blood vessels
- Ocular and cranial bruit
Subtle Signs
- Fever
- Nausea and vomiting
- Abnormal pulsations of the globe
- Nasal discharge of blood
Demographics
Septic thrombosis of the cavernous sinus is a potentially lethal disorder that often spreads from an infection of the face, mouth, throat, sinus, or orbit. Staphylococcal and streptococcal organisms are the most frequent cause. Most patients will have an elevated white blood count, and up to 70% will have positive blood cultures. Cerebrospinal fluid will indicate meningitis in 35%.
Mucormycosis is rarer and is seen in diabetic patients with ketoacidosis or in those with lymphoma or leukemia. Mortality may be as high as 90%.
Low-flow dural fistulas are usually spontaneous while high-flow is frequently caused by trauma. Dural fistulas close spontaneously or after carotid angiography nearly 50% of the time. High-flow fistulas rarely close spontaneously.
The frequency of Tolosa-Hunt’ syndrome is around 3% of all parasellar syndromes. It can range in patients from 3½ to 75 years, and there is no sex predilection, with the disease occurring more commonly in whites. Pain is a uniform finding.
About 2% of pituitary adenomas compress the intrinsic structures within the cavernous sinus. About 20% of nasopharyngeal tumors present as a cavernous sinus syndrome, and up to 20% of cavernous sinus syndromes are a result of malignant nasopharyngeal tumors. Up to 23% of tumors found in the cavernous sinus are metastatic.
Significant History
- Recent facial infection
- Sinusitis
- Diabetes
- Debilitated state
- Past history of cancer
- New onset facial or orbital pain
Ancillary Tests
Ocular assessment includes visual acuity; extraocular muscle testing; corneal sensitivity with cotton wisp; pupil evaluation; exophthalmometry; testing for any resistance to retropulsion; slit-lamp evaluation for arterialization of episcleral or conjunctival vessels; orbicularis muscle strength; and ocular, orbital, temple, and carotid auscultation. The patient’ temperature, nasal examination, and indirect pharyngoscopy and biopsies may be indicated.
Laboratory studies: CBC with differential, plasma glucose, ESR, antinuclear antibody, rheumatoid factor, chest x-ray, and neuroimaging of the sinuses, orbits, and brain. If infection is suspected, obtain blood cultures, cultures from the presumed primary source of infection, and lumbar puncture with cerebrospinal fluid analysis. If lymphadenopathy is present, a lymph node biopsy should be obtained. MRA or arteriography should be obtained in cases of suspected arteriovenous fistula.
The Treatment
Treatment depends on the source of the cavernous sinus syndrome. Infection will require hospitalization and intravenous antibiotics. Aseptic thrombosis may require systemic anticoagulation. Mucormycosis treatment includes amphotericin B and surgical debridement of necrotic tissue with stabilization of the metabolic state.
Treatment of a fistula is indicated for visual loss, intolerable pain, annoying tinnitus, or cosmesis. For fistulas of carotid origin, balloon embolization is used, while those of dural origin require particle embolization.
Intracavernous aneurysms are treated with balloon occlusion or surgical trapping.
Tolosa-Hunt’ syndrome responds quickly to oral prednisone, 80 to 100 mg once a day and is then tapered slowly as the pain subsides. Sometimes tapering may take months and recurrence is not uncommon.
Carotid Artery Dissection
Bruce G. Muchnick
ICD-9: 443.21
 THE DISEASE
THE DISEASE
Pathophysiology
Dissection represents a separation of the component layers of the carotid arterial wall because of the entrance and accumulation of intraluminal blood. Blood enters the vessel wall as a result of a breach in the endothelial layer. The resulting aneurysm may compromise the luminal diameter or cause an external diameter expansion. In either case, a second, false lumen is created of varying length. This second lumen may be stable, resolve, or occlude the true lumen, or burst through the entire wall and cause a severe hemorrhage. Subadventitial dissections cause aneurismal formation, whereas subintimal dissections cause mostly brain and eye ischemia. Subarachnoid hemorrhage may result from intracranial dissection.
Etiology
Dissecting carotid aneurysms may occur as a result of blunt trauma or penetrating injury of the head or neck. This includes head or neck surgery. Carotid artery dissection (CAD) may also arise spontaneously. Artery dissections are more common in individuals with predisposing systemic conditions such as connective tissue disease, Marfan’ syndrome, syphilis, atherosclerosis, migraine, temporal arteritis, and systemic hypertension. Mild trauma, such as jumping off a stepladder or treadmill running, has been shown to lead to persistent arterial wall dissection in the carotid artery.
The Patient
Clinical Symptoms
Ninety percent of all spontaneous dissections of the cervical internal carotid artery cause symptoms and 10% are silent and asymptomatic. Symptoms of dissection of carotid artery aneurysms are dependent on several factors, including the cause of the dissection, the site of the lesion, how the blood flow is affected, and whether aneurysmal rupture has occurred.
Minimally, and most commonly, there is headache (65% of cases) and facial (50%) or neck pain (15% of cases). If progressive thrombosis (occlusion) occurs, or distal embolization, then more complicated symptoms may occur because of cerebral or retinal infarcts. Neurological symptoms may include hemiparesis, aphasia, tinnitus, subjective bruit, transient ischemic attacks (TIA), monocular blindness, and diplopia. The symptom of double vision may arise from an upper cranial nerve palsy secondary to spontaneous dissection of the carotid artery. This occurs due to nerve ischemia from an interruption of blood flow secondary to the dissection of the carotid artery causing compression of the vasa nervorum.
Clinical Signs
The clinical signs of carotid artery dissection typically include neurological and visual deficits. These include the neurological signs of a stroke (including hemiparesis, tongue deviation, and declining consciousness), ocular or visual signs (including visual loss, scintillating scotomas, and Horner’ syndrome), and death. Horner’ syndrome is reported in about 35% of cases. Internal carotid artery dissection may lead to severe acute ocular ischemic syndrome. Cranial nerve palsies occur in 10% of cases, most commonly the lower nerves (IX to XII). Cranial nerve palsies affecting the extraocular muscles remain a rare sequella of carotid artery dissection.
Demographics
The incidence of artery dissection is unknown. In patients with completed stroke, the incidence of any artery dissection is 2.5%. In young individuals under 30 years of age with ischemic strokes, dissection is the cause in 22% of cases. About 1 in 3 patients with a dissecting artery have more than one involved vessel. The incidence of dissecting aneurysms is higher in men, most likely because of a higher rate of head or neck injury.
The peak age for a spontaneous carotid dissection is about 50 years of age. Seventy percent of patients are between the ages of 20 and 40 years of age. Atherosclerosis associated dissections average about age 59.
Half of patients with spontaneous carotid dissections have systemic hypertension. Ninety percent of patients with spontaneous carotid dissection present with a headache. Only 10% of patients with carotid dissection have eye, facial, or ear pain without headache.
Most patients who survive a spontaneous CAD do quite well, though about half report some residual neurological deficit. Traumatic CAD has a worse prognosis, often because of associated intracranial injury.
Significant History
- Does the patient have any sudden headache?
- Is there history of any head or neck trauma (no matter how trivial)?
- Is there history of any head or neck surgery?
- Is there history of TIAs?
- Is there history of neurological deficits?
- Does the patient have any sports-related head or neck injury?
- Does the patient have any history of systemic conditions that may contribute to the formation of CAD?
Ancillary Tests
Neuroimaging is used to confirm the presence of CAD in suspected patients.
- MR angiography
- Spinal CT angiography
- Conventional angiography
- Ultrasound (continuous wave Doppler ultrasound)
- Color Doppler flow imaging ultrasound
- Standard duplex ultrasound scanning
- Fat-saturated MRI cross sections
The Treatment
To treat the progressive thrombosis or distal embolization associated with CAD, medical treatment is directed at preventing further blood clotting. Immediate anticoagulation therapy is recommended in all patients with spontaneous dissection of the cervical portion of the internal carotid artery.
Standard anticoagulation, utilizing at first Heparin and later oral Coumadin, can reduce the risk of embolization, but may cause sudden, acute intracranial hemorrhaging. Rupture of a dissecting aneurysm remains a rare outcome of anticoagulation therapy.
The surgical approach to dissecting aneurysms depends on the location and extent of the lesion. Such surgical approaches manipulate the artery by ligation, wrapping, resecting, reconstruction, or bypassing of the involved area.
The recurrence rate of infarction or arterial dissections is very low.
Tension-Type Headache
Leonid Skorin Jr and Lawrence D. Robbins
ICD-9: 307.81
 THE DISEASE
THE DISEASE
Pathophysiology
The pain involved in tension-type headaches is best explained by the myofacial-central-vascular model. Input from the supraspinal control areas for muscle contraction is mediated by the central nervous system, specifically cortical, subcortical, and limbic mechanisms that cause the muscle to spasm. With prolonged periods of time of muscle contraction, tissue hypoxia results from small blood vessel compression. With tissue ischemia, there results the release of bradykinin, lactic acid, serotonin, and prostaglandins. This further results in muscle contraction and pain. Most patients with chronic tension-type headaches, that are more than mild, also suffer from migraines.
Etiology
Tension-type headache is thought to be partly caused by voluntary contraction of musculature, at times precipitated by stress and poor sleep that leads to inflammation and pain. Tension-type headache is a physical medical condition involving brain chemistry, as well as involving the cervical spine, the craniocervical junction, and the temporomandibular joint. Certain individuals do have a definite predisposition to these headaches. Approximately 50% of headache patients have an anxiety disorder, and 20% have either recurrent major depression or dysthymia. Bipolar illness is seen in 4% to 6% of headache patients.
Most patients with chronic tension-type headache have what is termed transformed migraine. They have a history of episodic migraine initially that has progressed to a chronic daily headache (CDH) over the years. CDH is seen in about 4% of the population and is defined as at least 15 headache days per month for at least 6 months. In 80% of cases, this process is associated with medication overuse and the development of rebound headaches. Episodic tension headaches are recurrent episodes of tension headache lasting minutes to days, but fewer than 15 per month or 180 d/y.
Tension-type headaches are bilateral headaches with a pressing or tightening quality with usually no ocular symptoms or signs.
Clinical Symptoms
The patient with tension-type headache will complain of a constant aching, pressure, or tightness that is felt in a band around the head (i.e., the “hatband” area) and down the back of the neck. Occasionally, the patient may complain of soreness, cramping, or a feeling like a vise around the head or a weight on the head. Pain is usually bilateral but is sometimes worse on one side. The patient may also complain of dizziness, nausea, photophobia, or phonophobia. CDH patients awaken with their headache and go to sleep with it.
Clinical Signs
Prominent Signs
- Tender spots along cranial and neck musculature
- Sharply localized nodules in the cervical or pericranial muscles
Demographics
Tension-type headache is the most common type of headache, with a lifetime incidence of 69% in men and 88% in women. Tension headaches account for 47% of the headaches treated in a primary care practice. These headaches may begin at any age, but in 40% of patients, they begin in childhood or adolescence. It is bilateral 90% of the time and strictly unilateral 10% of the time. The majority of patients with tension headache also experience periodic migraine headaches.
Significant History
- Myofascial pain syndrome
- Fibromyalgia
Ancillary Tests
A thorough headache history and complete ocular examination is necessary. Also, a complete neurologic evaluation should be performed. Muscles of the scalp and neck including the paraspinal and trapezius muscles should be palpated for tenderness. The temporomandibular area should be palpated to rule out dental occlusion problems. The temporal artery area should also be palpated to rule out temporal arteritis. With an unusual presentation or in those with new onset CDH, neuroimaging may be obtained. We are particularly worried about relatively new onset headaches.
The Treatment
Patients should be instructed to reduce precipitating factors and avoid stressful factors. Lifestyle changes should be reviewed. For selected patients, self-help books, relaxation exercises, or biofeedback are effective. Application of cold brings symptomatic relief in up to 70% of patients. Massage, physical therapy, and osteopathic manipulation can also be of benefit. Psychotherapy improves coping and is worthwhile even if it does not directly decrease head pain.
Symptomatic treatment consists of simple analgesics, such as acetaminophen, aspirin, or other nonsteroidals. Ibuprofen, 400 or 800 mg, or naproxen sodium, 275 to 550 mg, has been found to be very effective. Combination analgesic-sedative medications should be used cautiously because they have the potential to cause dependence. Midrin consists of a vasoconstrictor, isometheptene, combined with dichloraphenazone, a nonaddicting sedative, and 250 mg of acetaminophen. Midrin is primarily a migraine abortive agent, but it is effective for many patients with tension headache. A similar medication, MigraTen, is similar to Midrin, with caffeine, and no sedative. Both Midrin and MigraTen are available in generic.
A realistic goal in using preventive medication is to improve the headache situation by 50% to 90%, while attempting to minimize medicine use. Prophylactic treatment includes the use of tricyclic antidepressants, such as amitriptyline hydrochloride. Doses should be started low and increased slowly every 3 to 7 days until the maintenance dose is reached. Alternative antidepressants such as fluoxetine or duloxetine, and the antiseizure medications sodium valproate, topiramate, or gabapentin are somewhat beneficial in CDH.
Migraine Headache
Leonid Skorin Jr and Lawrence D. Robbins
ICD-9: 346.0—CLASSICAL MIGRAINE
ICD-9: 368.12—TRANSIENT VISUAL LOSS
ICD-9: 346.1—COMMON MIGRAINE
ICD-9: 346.2—VARIANTS OF MIGRAINE
ICD-9: 346.8—OTHER FORMS OF MIGRAINE
 THE DISEASE
THE DISEASE
Pathophysiology
The patient with migraine has an inherited susceptibility to headache. The expression of an altered migrainous threshold in headache-prone persons may depend on the balance between inhibitory and excitatory neurocircuits that are influenced by a complex interplay of exogenous and endogenous factors.
Migraine pain usually develops at a time when regional cerebral blood flow is reduced. In patients who have migraine with aura, there appears to be an initial decrease in volume of blood (oligemia) that originates from the occipital region and spreads anteriorly. This spreading oligemia may also relate to the spreading depression, which is a neurophysiological phenomenon that represents a front of intense neural activity followed by neural silence. This spreading depression may also trigger a response of the trigeminovascular sensory fibers, resulting in neurogenic inflammation. This process consists of vascular dilation, enhanced leakage, or extravasation of plasma proteins across the endothelium with subsequent edema formation in vessel walls and a complex chain of chemical and physiologic events that excite, sensitize, and lower the threshold of the trigeminal nociceptive terminals. With the stimulation of these sensory fibers, substance P, bradykinin, calcitonin gene–related peptide (CGRP), and serotonin are released, with serotonin being a key neurotransmitter in migraine. CGRP appears to be the most important mediator in migraine.
Etiology
Various exogenous and endogenous triggers can help precipitate a migraine attack. Dietary factors appear to play a role in as many as 25% of migraineurs. The direct-acting vasoactive substances that can cause diet-precipitated migraine include amines (tyramine, phenylethylamine), nitrates (food coloring and preservative), monosodium glutamate (food additive), and alcohol. Indirect-acting vasoactive substances include caffeine and nicotine. Just as certain foods and other substances can trigger a headache, hunger, and its resultant hypoglycemia from missed meals, eating inadequate meals, dieting, or fasting can all cause headaches.
Prescription drugs can also induce a migraine attack. These include nitroglycerin, reserpine, nonsteroidal anti-inflammatory agents such as indomethacin, H2-receptor antagonists such as cimetidine and ranitidine, antibiotics such as griseofulvin and trimethoprim-sulfamethoxazole, and hormonal preparations such as oral contraceptives and estrogens.
The Patient
Migraine is an episodic headache syndrome lasting 4 to 72 hours and is characterized by recurrent attacks that vary widely in intensity, frequency, and duration. Status migrainosis applies to migraine headaches that exceed 72 hours in length. Symptoms and signs depend on migraine type.
Migraine Without Aura
Clinical Symptoms
- Unilateral, pulsing, throbbing, or pounding headache of moderate to severe intensity
- Can be aggravated by physical activity
- Accompanied by nausea and/or vomiting and photophobia or phonophobia
Clinical Signs
- Normal physical and neurologic exam
- Pallor of the face
- In women, there is often a positive relationship with menses
Migraine with Aura
Clinical Symptoms
Approximately 20% of migraineurs experience an aura that lasts 10 to 20 minutes. The most common aura is visual and includes teichopsia, fortification spectrum, scintillating scotomas, and photopsias. Other aura include tingling (paresthesias) or numbness, motor disturbances such as monoparesis or hemiparesis, and cognitive or language disorders. Headache follows the aura. Vertigo or ataxia may occur.
Clinical Signs
- Normal physical and neurologic exam except for transient neuroparesis or hemiparesis
Migraine Aura Without Headache (Acephalgic Migraine)
Clinical Symptoms
- Same as migraine with aura but no headache
Clinical Signs
- Same as migraine with aura
Complicated Migraine
Prodromal neurologic manifestations last for the entire headache attack or may even leave a permanent defect.
- Ophthalmoplegic migraine—The third cranial nerve is affected most often in children.
- Basilar migraine—Visual symptoms are similar to those of migraine with aura but affect both visual fields. Also will see vertigo, diplopia, disturbed consciousness, tinnitus, and ataxia.
- Hemiplegic migraine—Characterized by both motor and sensory symptoms occurring in a unilateral distribution.
- Retinal migraine—Fully reversible monocular scotoma or blindness lasting less than 60 minutes.
Demographics
Migraine is a common condition with a prevalence of 18% in females and 7% in males. Migraine without aura accounts for 80% to 85% of all vascular headaches. The peak ages are between 20 and 35 years old. Twenty-five to forty percent of women with migraine suffer one to four or more severe, disabling attacks per month. Up to 75% of migraine patients report a first-degree relative having had migraine.
Significant History
Specific pattern of headache and the presence or absence of certain associated signs and symptoms.
Ocular assessment includes visual acuity, extraocular muscle testing, pupil testing, intraocular pressure, fundus evaluation, and visual fields. A complete physical and neurologic examination is also necessary. Additional tests that are generally useful for diagnosis of headache are MRI of the brain, MRA to rule out intracranial aneurysms, lumbar puncture, and CT scan of the sinuses. Whether to do any additional testing at all depends upon the clinician’ suspicion of organic pathology. Situations that raise the concern about organic pathology include
1. Progressive headaches over days or weeks, increasing in intensity
2. New onset headaches, particularly in patients who never have headaches
3. New onset headaches with exertion
4. Neurologic symptoms or signs, stiff neck, papilledema, and changes in level of consciousness
5. Fever that is not explained
6. Radical increase or change in a preexisting headache pattern
The Treatment
Nonpharmacologic treatment includes avoiding potential triggering factors, stress control, biofeedback, relaxation techniques, and cold application. Maintaining a headache calendar to better understand one’ new onset headache is often helpful (Table 19-1). Symptomatic treatment includes simple analgesics such as aspirin or acetaminophen or nonsteroidal anti-inflammatory agents, such as ibuprofen or naproxen sodium, for mild to moderate headaches and combination analgesics, such as isometheptene with acetaminophen and dichloralphenazone (Midrin), or a similar form with caffeine (MigraTen). Stronger symptomatic medications include corticosteroids, dihydroergotamine (available in various forms), opioid analgesics, or serotonin agonists such as sumatriptan. The serotonin agonists (triptans) are available in subcutaneous injection, tablet, nasal spray, and dissolving sublingual tablet form. Triptans have become the preferred first-line treatment of choice. Triptans are more efficacious when used early in a migraine.
TABLE 19-1 Headache Calendar for Patients

Appendix C. Headache Calendar for Patients. (Adapted from Robbins LD. Management of Headache and Headache Medications. 2nd ed. New York: Springer-Verlag, 2000:263–264, with permission.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


