 Neural Tumors of the Eyelid
Neural Tumors of the EyelidEyelid Neurofibroma
General Considerations
Neurofibroma is an important neural tumor that can affect skin in all parts of the body. It can develop as a solitary lesion unassociated with systemic disease or as multifocal or diffuse lesions associated with type 1 neurofibromatosis. The eyelid and orbit can be involved by neurofibroma in three different ways: solitary neurofibroma, multiple localized neurofibromas, and plexiform neurofibroma (1,2,3,4,5,6,7,8,9,10,11). Plexiform neurofibroma of the eyelid characteristically extends for some distance into the orbit.
Clinical Features
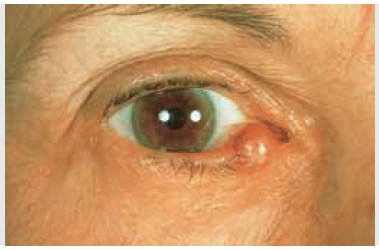
Solitary neurofibroma, also called fibroma molluscum, is a circumscribed subcutaneous nodular lesion of variable size. It may sometimes be painful, reflecting its peripheral nerve origin. Solitary neurofibroma can occur on the eyelid in patients who have no apparent neurofibromatosis. Such a lesion can initially resemble a chalazion or malignant eyelid tumor.
Multiple neurofibromas that affect the skin in patients with type 1 neurofibromatosis can also simultaneously occur on the eyelids (1,2,3,5,6). They appear as multiple, discrete, subcutaneous nodules on the eyelid and adjacent skin. They tend to be stable, but can sometimes gradually enlarge. Like the similar neurofibromas on the extraocular skin, they probably have a low potential to undergo malignant transformation; malignant peripheral nerve sheath tumors in the eyelid are quite rare.
Plexiform neurofibroma is considered pathognomonic of type 1 neurofibromatosis (von Recklinghausen’s disease). Eyelid involvement is usually associated with contiguous involvement of the deeper tissues in the orbit (1,2,3,4). It develops in young children as a thickening of the entire eyelid that initially causes an S-shaped curve to the margin of the upper eyelid (1,2,3,4). This lesion can show gradual progressive enlargement and extend into the malar area of the eyebrow and conjunctiva.
Pathology
In contrast to schwannoma (discussed in the next section), which is a tumor composed almost exclusively of Schwann cells, neurofibroma is composed of a combination of Schwann cells, peripheral nerve axons, endoneural fibroblasts, and perineural cells. There are several variations of cutaneous neurofibromas (7). In brief, each tumor type is composed of bundles of enlarged peripheral nerves. Bodian stain helps to delineate the axons and additional special stains can assist in the diagnosis and in identification of elements of the tumor (7).
Management
A small, asymptomatic solitary neurofibroma of the eyelid can be followed conservatively. Surgical resection can be withheld until there is significant growth or pain develops. If surgery becomes necessary, the lesion can usually be removed completely by way of an eyelid crease incision; wide margins are not necessary. If the overlying skin is spared, it can be preserved, facilitating a primary closure. The multiple small cutaneous neurofibromas associated with neurofibromatosis are usually asymptomatic and can be followed without active treatment. Larger lesions may be individually resected.
Diffuse or plexiform neurofibroma may be much more difficult to manage. The lesion is often ill-defined and bleeds profusely at the time of surgical resection (9,10,11). When such a lesion becomes cosmetically unacceptable, surgical debulking may be attempted (8). The carbon dioxide laser is reported to be of great assistance in such cases (9,10). Because of the extent of many plexiform neurofibromas, a multidisciplinary approach with ophthalmologists, otolaryngologists, and neurosurgeons is often necessary.
Selected References
1. Shields JA, Shields CL. The systemic hamartomatoses (“Phakomatoses”). In: Mannis MJ, Macsai MS, Huntley AC, eds. Eye and Skin Disease. New York: Lippincott-Raven; 1996:367-380.
2. Shields JA, Shields CL. The systemic hamartomatoses (“Phakomatoses”). In: Shields JA, Shields CL, eds. Intraocular Tumors. A Text and Atlas. Philadelphia: WB Saunders; 1992:520.
3. Shields JA. Peripheral nerve tumors of the orbit. In: Shields JA, ed. Diagnosis and Management of Orbital Tumors. Philadelphia: WB Saunders; 1989:149-169.
4. Brownstein S, Little JM. Ocular neurofibromatosis. Ophthalmology 1983; 90:1595-1599.
5. Lewis RA, Riccardi VM. von Recklinghausen neurofibromatosis. Ophthalmology 1981;88:348-354.
6. Farris SR, Grove AS Jr. Orbital and eyelid manifestations of neurofibromatosis: a clinical study and literature review. Ophthal Plast Reconstr Surg 1996;12:245-259.
7. Reed RJ, Argenyi Z. Tumors of neural tissue. In: Elder D, Elenitsas R, Jaworsky C, et al., eds. Lever’s Histopathology of the Skin. Philadelphia: Lippincott-Raven; 1997:980-982.
8. Lee V, Ragge NK, Collin JR. Orbitotemporal neurofibromatosis. Clinical features and surgical management. Ophthalmology 2004;111:382-388.
9. Lapid-Gortzak R, Lapid O, Monos T, et al. CO2 laser in the removal of a plexiform neurofibroma for the eyelid. Ophthalmic Surg Lasers 2000;31:4324.
10. Dailey RA, Sullivan SA, Wobig JL. Surgical debulking of eyelid and anterior orbital plexiform neurofibromas by means of the carbon dioxide laser. Am J Ophthalmol 2000;130:117-119.
11. Marchac D, Britto JA. Remodeling the upper eyelid in the management of orbitopalpebralneurofibromatosis. Br J Plast Surg 2005;58:944-956.
Eyelid Neurofibroma: Localized and Plexiform Types
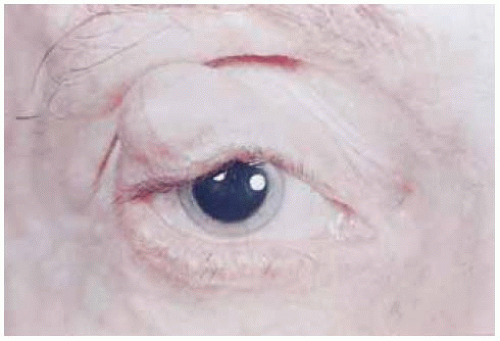 Figure 7.1. Solitary neurofibroma (fibroma molluscum) of the upper eyelid in a patient without neurofibromatosis. |
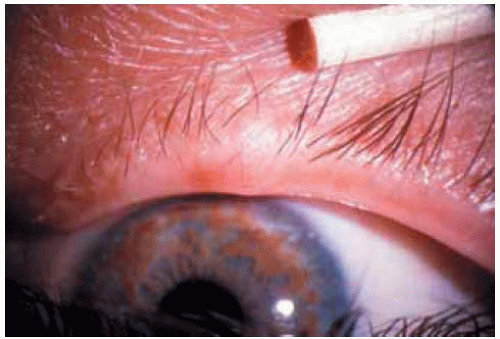 Figure 7.2. Small neurofibroma of upper eyelid in patient with neurofibromatosis type 1. (Courtesy of Douglas Cameron, MD.) |
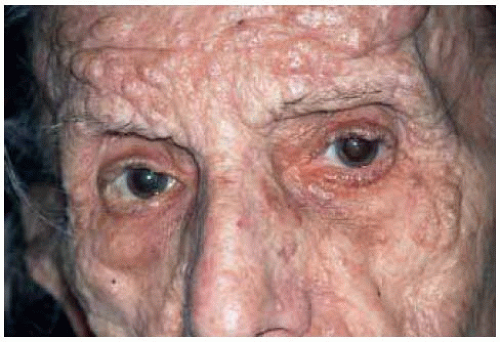 Figure 7.3. Facial appearance of multiple peripheral nerve sheath tumors (neurofibromas) in an 81-year-old woman with von Reckling-hausen’s neurofibromatosis. |
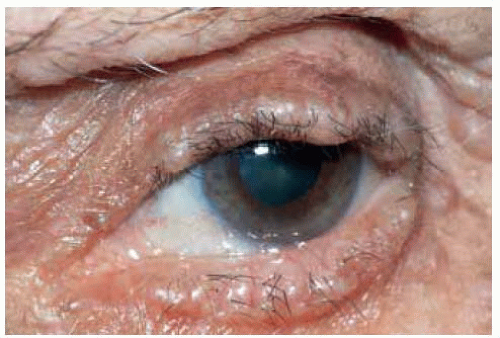 Figure 7.4. Closer view of eyelid margin of patient shown in Figure 7.3, demonstrating the multiple eyelid nodules. |
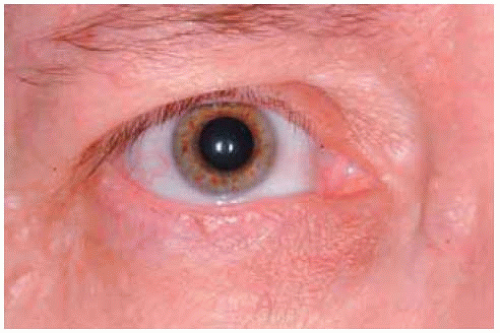 Figure 7.5. Multiple subtle localized neurofibromas in a patient with neurofibromatosis. The most prominent ones are just below the eyebrow superotemporally, immediately above the cilia superonasally, and on the lower eyelid near the lateral canthus. Note the characteristic Lisch nodules on the iris. |
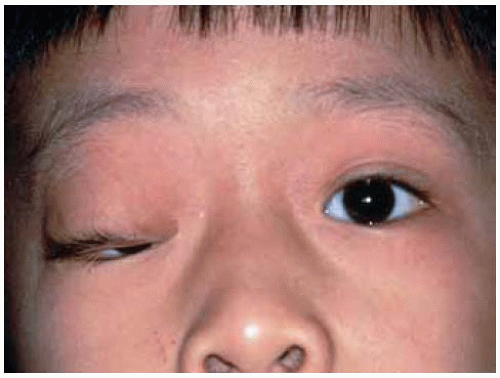 Figure 7.6. Plexiform neurofibroma of upper eyelid and anterior orbit in a 6-year-old boy with von Recklinghausen’s neurofibromatosis. Note the eyelid thickening and secondary blepharoptosis. |
Eyelid Neurofibroma: Plexiform Type
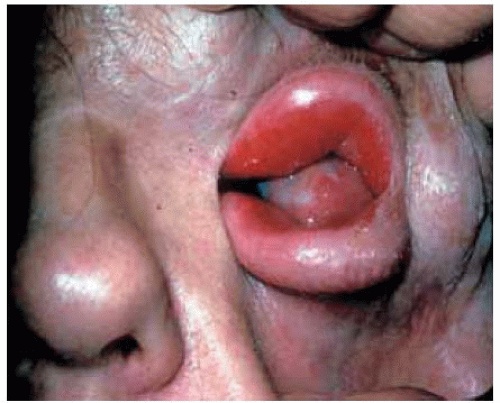 Figure 7.7. Massive plexiform neurofibroma of upper and lower eyelids in a patient with type 1 neurofibromatosis. Note the contiguous conjunctival and corneal neurofibroma. (Courtesy of Douglas Cameron, MD.) |
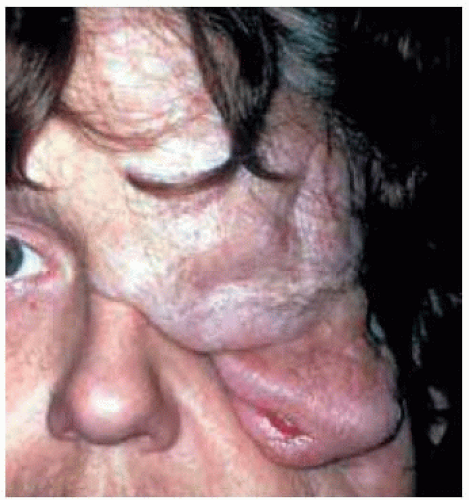 Figure 7.8. Massive plexiform neurofibroma of left side of face, with eyebrow and eyelid involvement. (Courtesy of Douglas Cameron, MD.) |
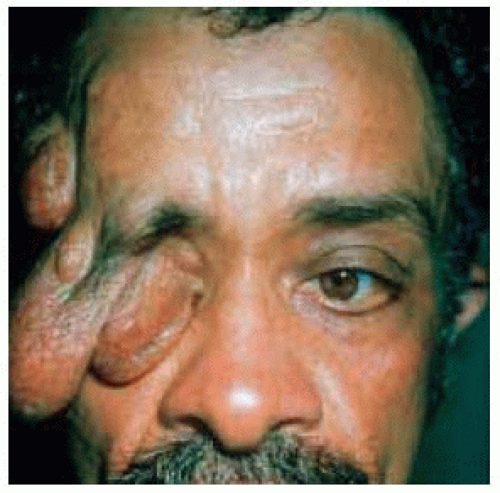 Figure 7.9. Massive plexiform neurofibroma of upper eyelid in a patient with neurofibromatosis. (Courtesy of Charles Lee, MD.) |
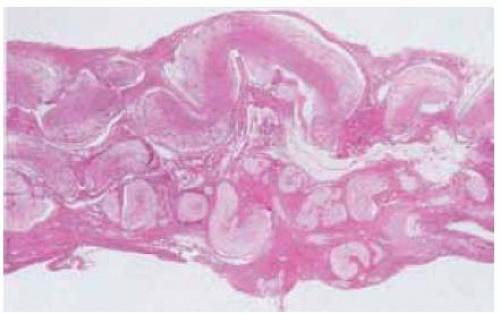 Figure 7.10. Low-power photomicrograph showing intertwining enlarged nerves typical of plexiform neurofibroma. (Hematoxylin-eosin 5.) |
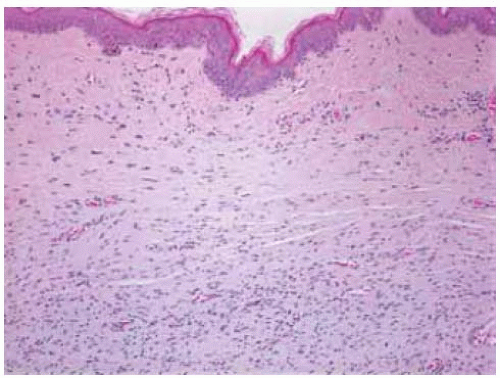 Figure 7.11. Pathology of cutaneous neurofibroma showing low-grade spindle cells. (Hematoxylin-eosin 15.) |
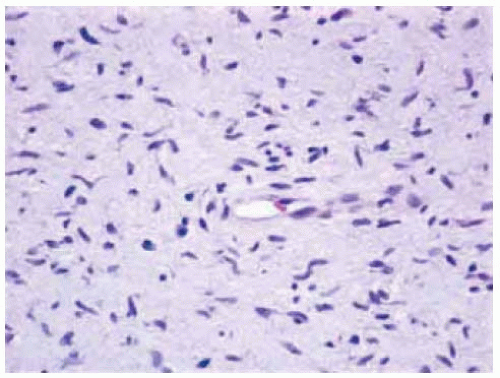 Figure 7.12. Photomicrograph of cutaneous neurofibroma showing randomly arranged spindle cells with mucinous cytoplasm. (Hematoxylin-eosin 25.) |
Eyelid Schwannoma (Neurilemoma)
General Considerations
Schwannoma (neurilemoma) is a benign peripheral nerve sheath tumor that is composed almost exclusively of a proliferation of Schwann cells. Multiple schwannomas can occur in patients with neurofibromatosis, but solitary schwannoma is usually unassociated with that entity. Schwannoma is known to arise in the orbit (1,2,3,4,5,6,7,8); occasionally, it occurs in the uveal tract, conjunctiva, caruncle, or eyelid. Only a few cases confined to the eyelid have been reported (3,4,5,6,7,8). Eyelid cases can occur in elderly patients and young children.
Clinical Features
Clinically, schwannoma of the eyelid appears as a firm subcutaneous mass that can simulate a chalazion. However, it generally is nonpainful, lacks inflammation, and grows very slowly.
Pathology
Histopathologically, schwannoma is an encapsulated lesion that is composed of closely compact spindle cells (Antoni A pattern) and larger, more round clear cells (Antoni B pattern). Most tumors display a combination of the two patterns.
Management
Management is complete excision; incomplete removal is associated with eventual recurrence and more aggressive behavior (3,4,5).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


