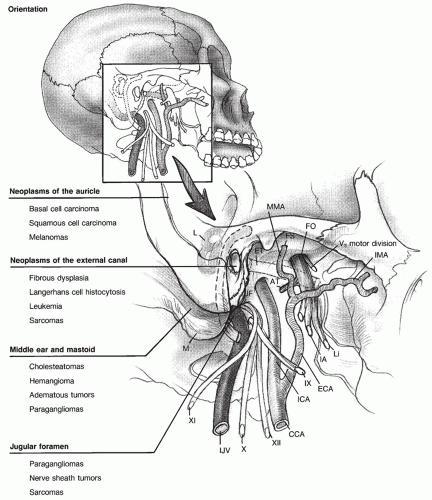
Paraganglioma
Paragangliomas are the most common true neoplasm of the middle ear and are considered the most frequent pathologic condition involving the jugular foramen. Paragangliomas are generally slow-growing, benign neoplasms with an interval between initial symptoms and diagnosis measured in years, and an estimated doubling time of 4.2 years. The growth patterns of temporal bone paragangliomas often follow the path of least resistance and are therefore greatly influenced by the site of tumor origin and regional anatomy. Paragangliomas that originate in the middle ear are termed glomus tympanicum tumors. Paragangliomas that originate in association in the jugular fossa are termed glomus jugulare tumors. Clinical manifestations are related to tumor extent and vascularity, as are treatment options.
Paragangliomas of the temporal bone arise from glomus bodies occupying the adventitia of the jugular bulb, along the course of the tympanic branch of the glossopharyngeal nerve (Jacobson nerve) or the auricular branch of the vagus nerve (Arnold nerve). Glomus bodies are groups of chemoreceptor cells belonging to the diffuse neuroendocrine system that are found in collections within the jugular foramen and middle ear, carotid bodies, the adrenal medulla, and along the aorta and vagus nerve. Histologically, they are identical to the carotid body and are similar to the autonomic ganglia of the adrenal medulla. Glomus bodies consist of clusters of chief cells supplied by a network of arterioles, venules, and both afferent and efferent nerve terminals. They are of neural crest derivation, migrating during embryogenesis to concentrate around autonomic ganglia. They are more accurately referred to as paraganglia, because they appear to play a role as neuromodulators or monitors of vascular activity. Chief cells have neurosecretory granules that contain norepinephrine, dopamine, and epinephrine, suggesting that the release of granule contents into the vascular system helps to regulate cardiorespiratory function, modify local blood distribution, and maintain thermoregulation. Unlike the carotid body and the adrenal medulla, however, paraganglia of the temporal bone play an uncertain role in these neuroendocrine functions. Paraganglia of the temporal bone are distinguished from other components of the diffuse neuroendocrine system, such as the adrenal medulla, by their lack of affinity for chromium salts used in certain histologic stains. They
are therefore categorized as nonchromaffin paraganglia. Adult temporal bones usually have only two or three paraganglia, but on occasion, there may be more, particularly during the fifth decade of life. Most paraganglia of the temporal bone are found in the anterolateral region of the jugular fossa and within the middle ear. Neoplastic transformation of paraganglia can occur in either location, indicated by local tumor invasion or metastatic spread. Metastases of head and neck paraganglioma are uncommon and are more frequently associated with non-carotid body paragangliomas of the head and neck. They occur in only 0.016% cases of paragangliomas and are most often found in the cervical lymph nodes, followed by lungs, liver, spleen, and bone (
1).
Paragangliomas represent 0.6% of all head and neck tumors, with a reported incidence range of 1 in 30,000 to 100,000. They occur more frequently on the left side, and females are affected by paragangliomas more frequently than males, 4-6:1. Living at high altitude is also a risk factor. Although the peak incidence occurs during the fifth decade of life, paragangliomas may present from infancy to old age. Tumors that arise in very young patients should generate additional concern, because they tend to be more aggressive, are more commonly multifocal, and are more likely to secrete vasoactive substances (
2). Approximately 5% to 10% of patients with paragangliomas eventually present with multiple tumors, but when paragangliomas occur as a familial form of an autosomal dominant disorder (about 10% of patients), multicentricity may occur in as many as 78% (
3). Mutations of loci encoding succinate dehydrogenase have been implicated in the development of familial paragangliomas (
4). Succinate dehydrogenase is an intracellular enzyme that plays a critical role in cellular energy production, and if the enzyme is inactive, succinate is allowed to accumulate, producing pseudohypoxic conditions that inhibit some neuroendocrine apoptotic factors. This may be the trigger that leads to proliferation of paraganglia.
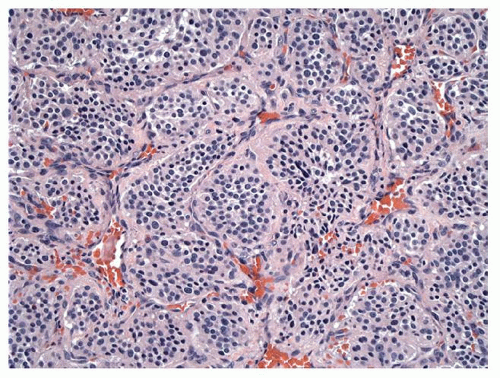
On gross examination, paragangliomas are deep red, firm, rubbery masses that bleed profusely upon manipulation. The histologic appearance of paraganglioma is distinctive. Nests of argyrophilic chief cells, termed
zellballen, are septated by a prominent fibrovascular stroma, though this characteristic appearance is occasionally not evident in temporal bone paragangliomas (
Fig. 147.2). Unmyelinated nerve fibers may be identified, but they are scarce when compared with normal paraganglia. Nuclear pleomorphism and hyperchromatism are prominent in chief cells but do not appear to indicate malignant growth. Electron microscopy reveals neurosecretory granules within chief cells that store catecholamines. Variable cranial nerve invasion may occur, a feature that has significance during surgical removal of the tumor mass.
Glomus tympanicum tumors usually originate on the promontory of the cochlea. As they grow, they follow the path of least resistance. First, the tumor enlarges to fill the middle ear and to envelope the ossicles. Patients present at this stage with conductive hearing loss and
pulsatile tinnitus caused by direct transmission of vascular pulsations from the highly vascularized tumor to the ossicles. Pulsatile tinnitus is the most common presenting symptom for patients with temporal bone paragangliomas. Because glomus tympanicum tumors enlarge within the middle ear cavity, patients with these tumors generally present with pulsatile tinnitus at an earlier stage than patients with glomus jugulare tumors. The tympanic membrane often remains intact as a glomus tympanicum tumor grows, but the tumor may displace the membrane laterally. If tumor extends through the tympanic membrane into the external auditory canal, patients may present with otalgia or bloody otorrhea. As glomus tympanicum tumors enlarge further, they may extend into the mastoid antrum
via the aditus ad antrum, into the facial recess, or into the retrofacial air cell tract. At this stage, the tympanic and mastoid portions of the facial nerve may become involved with tumor. The tumor may grow anteriorly into the eustachian tube and inferiorly into the infralabyrinthine air cell tract. When the tumor causes bone erosion in the hypotympanum, the jugular fossa or the vertical portion of the petrous carotid artery may be exposed. It is difficult to distinguish an extensive glomus tympanicum tumor of this kind from a glomus jugulare tumor. Patients with extensive tumors may present with multiple cranial nerve neuropathies.
Glomus jugulare tumors arise in the jugular fossa and are usually large before patients become symptomatic. These are more likely to secrete catecholamines than the glomus tympanicum variety. Compression of neurovascular structures in the jugular fossa and extension medially along the skull base to the hypoglossal canal can lead to cranial nerve neuropathies manifesting as dysphagia, dysphonia, aspiration, and dysarthria. Erosion of the jugular fossa anteriorly and superiorly exposes the petrous carotid artery and allows tumor to invade the middle ear, causing conductive hearing loss and pulsatile tinnitus. Intracranial extension occurs when glomus tumors grow into the eustachian tube and extend into the peritubal air cell tract or follow the petrous carotid artery into the petrous apex, the cavernous sinus, and the middle cranial fossa, resulting in facial hypesthesia. Glomus jugulare tumors may involve the posterior cranial fossa when they extend medially along the skull base or through the infralabyrinthine air cell tract. A patient with extensive tumor that compresses the cerebellum and the brainstem in the posterior fossa may present with ataxia and imbalance.
Paragangliomas of the temporal bone can usually be diagnosed on the basis of findings on physical exam and characteristic features found on imaging studies. Otoscopic examination of a middle ear paraganglioma frequently demonstrates a reddish-blue pulsatile mass medial to the inferior tympanic membrane. Positive pressure during pneumatic otoscopy causes blanching of the mass, a phenomenon known as the Brown sign. The pulsatile nature of the tumor can be diminished with ipsilateral carotid artery compression, a positive Aquino sign. Objective tinnitus may be apparent if auscultation over the mastoid or infra-auricular area reveals an audible bruit. When tumor extends through the tympanic membrane, otoscopic examination shows a hemorrhagic aural polyp. Tumors involving the jugular foramen can be identified when lower cranial nerve palsies develop. Jugular foramen syndrome, also termed Vernet syndrome, arises when tumor growth affects cranial nerves IX, X, and XI and causes paresis or paralysis of the muscles innervated by these nerves. Villaret syndrome is a combination of jugular foramen syndrome with Horner syndrome in patients with more extensive disease. Patients with paragangliomas that erode the carotid canal and compromise the sympathetic plexus present with Horner syndrome (miosis, ptosis, anhidrosis, and enophthalmos). If facial nerve weakness or paralysis exists, it denotes extensive involvement of the middle ear and mastoid. Tuning fork tests or complete audiometric evaluation in these patients shows conductive hearing loss and on rare occasions sensorineural hearing loss. Ataxia or rostral cranial nerve palsies are disquieting signs that indicate involvement of the posterior cranial fossa or cavernous sinus.
Although the chief cells of paragangliomas have neurosecretory granules that store catecholamines, only 1% to 3% of these tumors actively secrete norepinephrine. Catecholamines are much more likely to be secreted by glomus jugulare tumors than by glomus tympanicum tumors. Opinions vary regarding the need to screen for functionally active temporal bone paraganglioma, but all tumor patients who present with a history of flushing, frequent diarrhea, palpitations, headaches, poorly controlled hypertension, orthostasis, or excessive perspiration should have serum catecholamine levels measured and 24-hour collection of urine for analysis of vanillylmandelic acid and metanephrine.
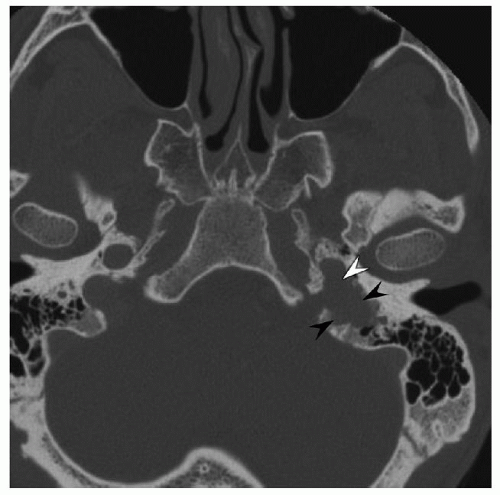
Diagnostic imaging studies provide essential information for the evaluation of patients with temporal bone paragangliomas. These studies reveal diagnostic facts and details about tumor extent and regional anatomy that are essential in planning surgery. High-resolution computed tomography (HRCT) of the temporal bone uses a thin-section bone algorithm and is usually the first imaging study ordered to evaluate a patient suspected of having a temporal bone paraganglioma. HRCT can identify the tumor origin accurately when the bony partition between the jugular fossa and the hypotympanum is intact. In this instance, a glomus jugulare tumor that erodes the jugular fossa and involves lower cranial nerves can be distinguished from a glomus tympanicum tumor that occupies the middle ear. Without this bony partition, it may be difficult to identify the origin of a temporal bone paraganglioma. Erosion of the caroticojugular spine that separates
the jugular bulb and the petrous carotid artery usually indicates a glomus jugulare tumor (
Fig. 147.3). If the spine is eroded and the carotid canal is exposed, involvement of the petrous carotid artery is likely. HRCT also helps to identify other lesions that should be excluded, such as a dehiscent jugular bulb or an aberrant internal carotid artery. Obliteration of the fallopian canal indicates adherence of tumor to the facial nerve or invasion of the nerve by tumor. Multidetector CT angiography can diagnose glomus tumors with accuracy and offers information about feeding vessels, venous drainage, and jugular involvement (
5). Intracranial extension can also be identified, but magnetic resonance imaging (MRI) is better than HRCT to evaluate the relationship between paraganglioma and adjacent soft tissue structures. MRI will not only identify intracranial extension, but images may help differentiate between intradural and extradural extension. MRI signal characteristics diagnostic for paragangliomas include vascular flow voids within the tumor, the so-called salt and pepper pattern. Pepper refers to the signal voids of large feeding arteries, and salt represents subacute hemorrhage within the tumor. Magnetic resonance angiography and magnetic resonance venography may demonstrate intraluminal involvement of the petrous carotid artery or occlusion of the jugular vein and sigmoid sinus. However, these studies are usually not useful for most large tumors as formal angiography will be necessary. MRI of the neck offers enough detail to screen for multicentric disease such as carotid body tumors or glomus vagale tumors, but other imaging modalities that exploit common features of neuroendocrine tissue may be useful in detecting tumors and recurrences. 123I-MIBG (metaiodobenzylguanidine) scintigraphy and (
18)F-DOPA positron emission tomography can be used to detect active tumors, but somatostatin receptor scintigraphy is more reliable for paragangliomas of the head and neck (
6).
The principal therapeutic modality is complete surgical excision of the tumor. Secreting tumors must be treated with adjunctive a and β blockade. If surgical therapy is deemed appropriate, small tumors isolated to the promontory can be successfully removed via the transcanal or hypotympanotomy approach. More advanced lesions of the middle ear and mastoid can be exposed with an extended facial recess approach through the mastoid. Large paragangliomas should be evaluated preoperatively with four-vessel angiography. Angiography is combined with embolization using polyvinyl alcohol or intravascular coils 1 or 2 days before surgery to decrease intraoperative blood loss, shorten duration of the procedure, and reduce postoperative morbidity. Devascularization of the entire tumor is difficult, requiring embolization of each vascular pedicle, and may prove impossible in tumors with intracranial extension (
7). Sacrifice of the internal carotid artery when invaded by tumor is seldom necessary, and thus, balloon occlusion studies are rarely performed in these patients. Instead, residual mural tumor and tumor that encases the internal carotid artery after subtotal resection is observed or treated with stereotactic radiation surgery (SRS). Glomus jugulare tumors are approached via transmastoid-transcervical exposure of the jugular bulb and jugular foramen. Large tumors require limited facial nerve rerouting or formal infratemporal fossa dissection through this approach. In some cases, more extensive temporal bone dissection is necessary to eradicate tumor that has spread to the petrous apex. This may necessitate removal of part or all of the labyrinth. Intracranial extension requires a combined neurosurgical-neurotologic procedure. Postoperative cranial nerve palsies are not uncommon, and some patients require follow-up care to assist with facial reanimation, deglutition, and phonation. Alternatives to surgical therapy for primary, recurrent, or persistent disease include external beam radiation therapy, intensity-modulated radiation therapy (IMRT), and SRS. Radiation has little effect on the primary tumor cells, the chief cells, but it causes obliterative endarteritis in tumor vessels that can stop tumor growth. One recent meta-analysis suggests that SRS provides better tumor control than surgery, with diminished risk to cranial nerves, but large tumors may require surgical debulking prior to radiosurgery (
8).
Epidermoid (Cholesteatoma)
Epidermoids are soft tissue masses resulting from an aberrant accumulation of keratin debris within a sac of squamous epithelium. They are customarily classified as tumors though they are not strictly cellular growths and therefore not neoplastic. Epidermoids may result from squamous epithelial entrapment during embryogenesis, mucosal metaplasia, or, most commonly, with anomalous squamous epithelial migration or deposition. The
nomenclature used to describe these masses is determined based on site of origin and pathogenesis. As an example, masses that arise from congenital epithelial rests in the petrous apex, internal auditory canal, or cerebellopontine angle are generally referred to as epidermoids. Cholesteatoma is the name for epidermoids resulting from the migration of tympanic membrane epithelial cells into the middle ear or from traumatic implantation deep to the skin of the external auditory canal. Congenital cholesteatoma, in contrast, usually refers to an epithelial sac in the middle ear arising from the entrapment of congenital epithelial rests during development. Despite diversity of origin and location, all possess similar histology and growth potential.
Middle ear cholesteatomas are generally divided into two types, congenital and acquired.
Congenital middle ear cholesteatoma occurs in the presence of an intact tympanic membrane, exclusive of any history of otorrhea, prior otologic intervention, or previous perforation. Theories advanced to explain the origin of these lesions include invagination or implantation into the middle ear, mucosal metaplasia, and epidermoid formation from retained epithelial cell rests. Though the debate persists, the latter is favored as the most likely. Histologic examination of fetal temporal bones has demonstrated epidermoid formation throughout the annular lateral wall region, not solely in the anterosuperior annular region of the tympanic cavity as previously described (
9,
10).
Acquired cholesteatomas arise from the transposition of keratinizing squamous epithelium into the middle ear, epitympanum, or mastoid and are often associated with perforation or retraction of the tympanic membrane. These latter are by far the more common of the two, likely resulting from eustachian tube dysfunction or deficiencies in tympanic membrane structure, or a combination. Together, these qualities promote formation of a retraction pocket in the tympanic membrane, trapping epithelial cells within and leading to the accumulation of keratin debris in the middle ear. Acquired aural cholesteatoma may extend to the petrous bone or to the cranial cavity, but most lesions of the petrous apex and the cerebellopontine angle are thought to be of congenital origin. Cholesteatoma of the external auditory canal could conceivably result from congenital rests of tissue trapped deep to the skin of the canal, but most canal wall cholesteatomas occur after traumatic implantation of epithelium subsequent to external trauma or otologic surgery. Patients with cholesteatoma of the middle ear and external auditory canal most often present with purulent otorrhea and conductive hearing loss, while patients with congenital cholesteatoma rarely present with otorrhea as the tympanic membrane is usually intact.
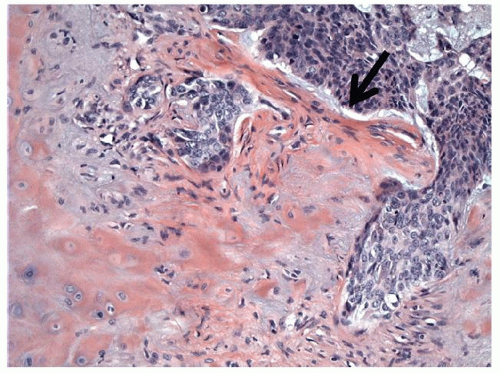
Epidermoids represent approximately 1% of all intracranial tumors, and most occur in the cerebellopontine angle. Squamous metaplasia within the temporal bone or intradural space has been proposed in the histogenesis of these lesions, but it is more likely that developmental entrapment of squamous epithelium from the neural tube is the cause. As squamous epithelium located at the periphery of an epidermoid comprises its only viable proliferating tissue, it grows very slowly in comparison with solid benign neoplasms of similar dimensions. Epidermoids enlarge by expanding to fill empty spaces such as the cerebellopontine angle, the internal auditory canal, or air cells that occupy the petrous apex. These masses are infiltrative, and they grow to a relatively large size before they begin to compress or displace adjacent structures, becoming clinically detectable. Additionally, epidermoids often incite a localized inflammatory reaction that causes the lining to become densely adherent to juxtaposed brainstem or cerebellum. Local neurovascular structures become enveloped by epidermoids instead of being displaced or compressed, and neurologic function may be altered when the vascular supply of a nerve is compromised by the infiltrative process. Most patients become symptomatic in a very gradual manner, their symptoms related to the location of the mass. Patients with internal auditory canal or cerebellopontine angle epidermoids often present with imbalance and hearing loss akin to patients with vestibular schwannoma. However, epidermoids are more likely than vestibular schwannoma to cause facial weakness or hemifacial spasm. Epidermoids are also more likely to compromise the trigeminal nerve causing diminished facial sensation or facial pain. Intracranial extension of these lesions may also manifest as diplopia or headache. All epidermoids and cholesteatomas exhibit a similar morphologic appearance. The friable lesions are either smooth and cystic with a round or oval appearance or nodular and irregular. The lining of the sac is usually white in color and spongy in consistency. Histologically, the cyst is lined with a benign keratinizing squamous epithelium consisting of three components: the sac or epithelial matrix, the perimatrix, and the contents of the cyst. Typical layers of squamous epithelium may be identified within the epithelial matrix. The contents of the cyst include fully differentiated laminated keratin. Acquired and congenital lesions can often be distinguished histologically by the thicker matrix and more vigorous proliferation of inflammatory cells within the sac and at the periphery of acquired lesions.
The diagnosis of cholesteatoma is usually made during otologic examination, while epidermoids are usually diagnosed with radiographic imaging studies. Congenital epidermoids are frequently identified as an asymptomatic mass in the anterosuperior quadrant of the middle ear. Patients with acquired disease have keratin debris, granulation polyps, or purulent material emanating from the mouth or opening of the sac. Clinical findings in patients with epidermoids include facial weakness or paralysis and sensorineural hearing loss when the lesion involves the internal auditory canal and/or the cerebellopontine angle. Facial hypesthesia and abducens nerve palsy occur when epidermoids invade the anterior petrous apex.
HRCT of the temporal bone in patients with epidermoid disease shows a well-defined homogeneous mass that occasionally contains areas of calcification or has eroded adjacent bony structures. MRI is diagnostic, revealing a well-circumscribed nonenhancing mass that has low signal intensity on T1-weighted images, high signal intensity on T2-weighted images, and hyperintensity on diffusionweighted image sequences.
Management of cholesteatomas of the external auditory canal and middle ear is addressed in other chapters in this text. Optimal treatment of epidermoids of the skull base consists of total microsurgical resection. This often necessitates a posterior fossa or middle fossa craniotomy, but transtemporal approaches may be indicated, especially in patients with nonserviceable hearing in the ipsilateral ear. Because the capsule of the mass may be densely adherent to the brainstem or intracranial vascular structures, complete removal of skull base epidermoids is extremely difficult or even impossible. Complete excision is achieved in only half of all patients with epidermoids, and additional postoperative cranial nerve deficits are discovered in most of these patients (
11). Recurrence can be expected in at least 30% of patients when subtotal removal is carried out. Malignant transformation is very rare, and only 26 cases have been reported.
Hemangioma and Hemangiopericytoma
Hemangiomas are benign vascular proliferations that arise from capillaries, arterioles, or venules. They are classified according to the type of vessel from which they originate: capillary hemangioma, cavernous hemangioma, and venous hemangioma. It is unclear whether they represent true neoplasms or hyperplastic growth of normal tissue occurring in an appropriate anatomic site. Hemangiomas are reported to occur in a variety of locations involving the external ear and lateral skull base, namely, the external auditory canal and tympanic membrane, the middle ear, the internal auditory canal, and the geniculate segment of the facial nerve. Tumors are spongy red or purple nodular masses. On microscopic examination, they show thin-walled vascular channels that contain blood and are small or intermediate in size. These channels are not surrounded by an elastic or muscular layer. Clinical presentation varies depending on tumor location. Hemangiomas of the external auditory canal and tympanic membrane have been reported when patients present with mild conductive hearing loss and aural fullness. Patients with middle ear tumors are often asymptomatic, but they can also present with conductive hearing loss, aural fullness, and pulsatile tinnitus. Otologic examination reveals a vascular intratympanic mass that can be confused with a paraganglioma, an adenomatous tumor, or an aberrant vascular anatomy. Preoperatively, hemangiomas of the internal auditory canal may be difficult to distinguish from vestibular schwannomas when patients present with unilateral sensorineural hearing loss. However, accompanying facial nerve dysfunction is more characteristic of hemangioma, even when tumors are small. CT imaging studies help to differentiate intracanalicular lesions when hemangiomas show calcium stippling, which is characteristic of an osseous hemangioma. On MRI, they are heterogeneously hyperintense.
Geniculate hemangiomas are the most common temporal bone hemangioma and perhaps the most intriguing. They consistently arise from the superior aspect of the geniculate ganglion, probably from vascular plexuses on the surface of the facial nerve, and extend into the floor of the middle cranial fossa. Even though the tumor is generally extraneural, they sometimes infiltrate the nerve or are associated with a localized inflammatory response that causes the tumor to adhere tightly to the nerve sheath. This intimate relationship with the facial nerve accounts for the frequently associated facial nerve dysfunction, manifest by paralysis, twitching, or spasm of the facial musculature, even when tumors are very small.
Depending on their size, location, and the structures they involve, hemangiomas of the ear and temporal bone are either observed or treated with complete surgical excision. Resection of lesions from the external canal and the middle ear is usually straightforward, but it may be unnecessary in pediatric patients who are asymptomatic as these lesions often involute spontaneously. When hemangioma is associated with the facial nerve, however, proper management is controversial. If facial paralysis exists, tumor resection with nerve grafting may be appropriate. If facial nerve function is normal or only mildly compromised, early excision when the tumor can be peeled off the nerve while preserving function may be appropriate. Facial nerve monitoring is used during the surgical resection to help maintain nerve integrity. When left intact, patients generally have superior results when compared to patients who require nerve sacrifice and grafting. In patients who have undergone subtotal resection for nerve preservation, regrowth has not been detected as late as 13 years after surgery. If a cochlear fistula is noted in the setting of serviceable hearing, surgery should be delayed in order to preserve hearing for as long as possible (
12). However, most case reports suggest that facial nerve integrity cannot be maintained when removing hemangiomas, and because most geniculate hemangiomas grow very slowly, observation may be the most appropriate management until severe dysfunction or facial paralysis occurs.
Hemangiopericytoma is a rare vascular tumor derived from pericytes that line the outside surface of capillary basal lamina. They can occur wherever capillaries are found. Less than 20% of hemangiopericytomas occur in the head and neck, and fewer than 10 cases have been reported where the tumor originated in the temporal bone. The middle ear, jugular fossa, and petrous bone were the apparent sites of origin of the tumors that occurred in the temporal bone. Symptoms include hearing loss, otorrhea, and cranial nerve deficits. Hemangiopericytoma can
occur in patients of any age with 10% of cases occurring in children, but it is most frequent in patients aged 50 to 70, affecting males and females equally. Tumors are soft or rubbery and pale gray or white in color. The metastatic rate for primary temporal bone tumors is estimated to be 15% to 20%. On microscopic examination, hemangiopericytomas are circumscribed pseudoencapsulated cellular tumors containing thin-walled vascular spaces separated by sheets of polyhedral- and spindle-shaped cells. They do not demonstrate intratumoral calcification. Tumors with malignant behavior may display nuclear pleomorphism, lymphocytic infiltration, and lack of vascular spaces with necrosis and hemorrhage. Wide excision with or without embolization is the treatment of choice, but external beam radiation therapy, stereotactic radiosurgery, or chemotherapy may be used to treat extensive, recurrent, or inoperable tumors. Long follow-up is mandatory secondary to their propensity to recur and metastasize (
13).
Lymphoma, Plasmacytoma, and Leukemia
Lymphoma is a neoplasm of the lymphoreticular system that may involve the temporal bone either as a secondary lesion after metastatic spread or as a primary lesion causing focal disease. Neoplastic infiltration of the middle ear, the facial nerve, the eighth cranial nerve, and the bone marrow of the petrous apex is not an uncommon finding during postmortem examination of temporal bones from patients with systemic lymphoma. However, most patients are asymptomatic unless lymphomatous infiltration results in middle ear effusion or hemorrhage and conductive hearing loss. Primary osseous lymphoma represents 1% to 2% of malignant lymphoma, and only 18 cases of primary temporal bone B-cell lymphoma have been reported (
14). In these case reports, the most common presenting symptoms were conductive hearing loss, otalgia, aural fullness, and otorrhea. Localized swelling and pain, fevers, sensorineural hearing loss, balance disturbance, and facial and abducens palsies from perineural invasion have also been reported. Physical examination may reveal middle ear masses or effusions and facial paresis. Once diffuse lymphoma has been ruled out, most patients with primary lymphoma of the temporal bone do well after chemotherapy and/or radiation therapy. The role of surgery in these patients is to obtain tissue for diagnosis.
Extramedullary plasmacytoma of the temporal bone is a rare neoplasm that is thought to arise as a clone of malignant plasma cells in the submucosal stroma of the middle ear. These are solitary lesions that occur outside the bony medullary spaces, and they are less likely to develop into disseminated disease or into multiple myeloma when compared with solitary osseous plasmacytomas. The majority of extramedullary plasmacytomas develop in the upper aerodigestive tract with only 1% of these tumors arising in the temporal bone. Patients, more often men over 50, present with aural fullness or otalgia, hearing loss, and tinnitus, and physical examination reveals a thickened tympanic membrane with an intratympanic mass or an aural polyp. Microscopic examination of these fleshy masses reveals sheets of monotonous round cells that are typical of plasma cells. Nuclear and cellular atypia is variable and defines tumor differentiation and grade. Tumor grade, however, is not necessarily predictive of tumor behavior. When biopsy specimens suggest the diagnosis of extramedullary plasmacytoma, patients should be evaluated for disseminated disease or multiple. This evaluation includes complete blood count with differential and smear, erythrocyte sedimentation rate, serum chemistries, creatinine, serum and urine protein analysis for monoclonal antibodies, bone marrow biopsy, and a radiographic skeletal survey. Chemotherapy is indicated for disseminated disease, but extramedullary plasmacytoma isolated to the temporal bone is usually successfully treated with external beam radiation therapy and surgery for radioresistant or very large tumors. Recurrence is rare as is dissemination to multiple myeloma, but the possibility necessitates long-term follow-up (
15).
Leukemia may involve the temporal bone by infiltrating marrow spaces and the tympanomastoid cavity or by causing hemorrhage within the middle or inner ear. In leukemia patients, marrow spaces in the temporal bone are frequently infiltrated by leukemic cells, but this rarely results in clinical manifestations. Infiltration of the middle ear and mastoid air cells is less common but may result in symptomatic effusions, often misdiagnosed as otomastoiditis, as these patients are particularly susceptible to infection. Patients with acute or chronic myelogenous leukemia may develop larger consolidated infiltrates that form solid tumors known as granulocytic sarcomas or chloromas. These present with hearing loss, facial palsy, and postauricular and auditory canal swelling and are often first mistakenly treated for infection. The tumors consist of immature granulocytes and contain myeloperoxidase that gives the lesion a green hue, hence the name chloroma. Most chloromas occur in children with leukemia. Leukemic infiltrates can involve the cochlea causing sensorineural hearing loss, but inner ear injury more likely results from local hemorrhage. Diagnosis is best made with biopsy, though this is often not feasible, and MRI is the imaging modality of choice. Treatment of systemic leukemia that infiltrates the temporal bone requires appropriate high-dose chemotherapy and management by a hematologist/oncologist with consideration for radiation therapy. Prognosis is poor, especially in the setting of delayed diagnosis (
16).