Metabolic
Alex V. Levin
Thomas W. Wilson
J. Raymond Buncic
Diseases of metabolism are classified according to the type of substance accumulated or absent due to an error in enzyme production or function, including amino acids (cystinosis, homocystinuria, phenylketonuria, and tyrosinemia), organic acids (glutaric aciduria and alkaptonuria), cholesterol (Smith-Lemli-Opitz), sugars (diabetes mellitus, galactosemia, and carbohydrate-deficient glycoprotein syndrome), lipids (abetalipoproteinemia, hyperlipoproteinemia, and lecithin acyltransferase deficiency), neural components (Fabry, galactosialidosis, mucopolysaccharidosis, mucolipidosis, and sphingolipidosis), peroxisomal (Zellweger, neonatal adrenoleukodystrophy, Refsum, and hyperoxaluria), and metals (Menkes and Wilson disease).
Ocular abnormalities in the sphingolipidosis are caused by accumulation of products of metabolic degradation of cerebral tissue. A cherry red spot is seen in Tay-Sachs, Niemann-Pick, Sandhoff, Farber, metachromatic leukodystrophy, and GM1 gangliosidosis. Optic atrophy is a manifestation of metachromatic leukodystrophy, Krabbe, Zellweger, and all mucopolysaccharidoses. Corneal abnormalities are common in Fabry (corneal verticillata) and corneal cloudiness in Lowe, metachromatic leukodystrophy, Zellweger, mucolipidoses, and mucopolysaccharidoses.
Kayser-Fleischer rings can be located in the peripheral cornea of patients with the copper metabolism abnormality Wilson disease. Cataracts are common in diabetes mellitus, mannosidosis (spoke-wheel cataract), Lowe, Fabry (spoke-wheel cataract), and Niemann-Pick (brown anterior lens changes).
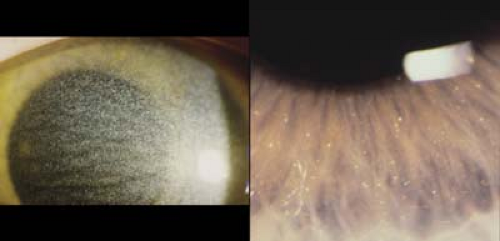 Figure 20.1 Cystinosis—Cornea Cystinosis is a group of metabolic disorders that results in excess cystine in the tissues throughout the body. Patients present with photophobia due to deposition of corneal crystals (left image). The crystals are deposited in the peripheral cornea and then progress centrally. Their highest concentration is in the anterior stroma. Patients can also have deposition of cysteine in other ocular tissues, and crystals have been observed in the anterior lens capsule, iris (right image), and retina. Oral cysteamine can decrease extracellular levels of cysteine. Clinical trials are currently in progress to determine the effect of cysteamine applied topically to decrease the corneal crystals. Renal failure is the major cause of nonocular morbidity. |
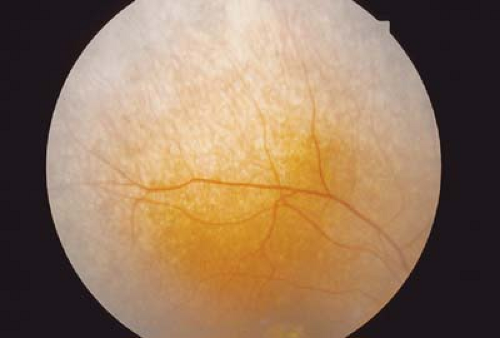 Figure 20.2 Cystinosis—Retina Pigment retinopathy is concentrated in the periphery and is characterized by pigment clumps and areas of atrophy. The macula may also be involved. Retinal crystals of cystinosis can be deposited with the layers of the retina. Cystinosis can lead to retinal dysfunction as demonstrated on electroretinogram and dark adaptation. Progressive vision loss is common. Systemic manifestations include renal failure from a deposition of cystine. Patients will often present with dehydration because of the decreased reabsorption of water. The deposition of cystine within body tissue confirms the diagnosis of cystinosis. Treatment of renal failure requires transplantation. |
 Figure 20.3 Homocystinuria Homocystinuria involves the abnormal metabolism of sulfur-containing amino acids. The most common enzyme deficiency is cystathionine β-synthase. Homocystinuria is inherited as an autosomal recessive trait and has been linked to chromosome 21. Patients will have increased levels of homocysteine and methionine. Ocular features include ectopia lentis and retinal detachment. Patients with homocystinuria are at increased risk of developing thromboembolic events and vascular occlusions. Patients often have skeletal changes similar to Marfan syndrome (arachnodactyly, scoliosis, and increased limb length [Marfanoid habitus]) but also often have mental retardation of learning delays not seen in Marfan syndrome. Treatment includes supplemental pyridoxine and cystine and a methionine-free diet. |
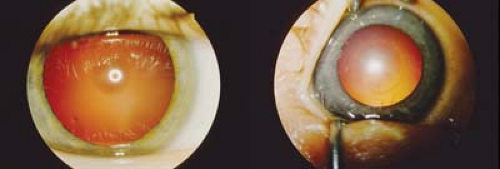 Figure 20.4 Galactosemia Galactosemia is a group of metabolic disorders with defects in galactose metabolism. Different enzyme deficiencies will lead to specific clinical features. Newborns with galactose-1-phosphate uridylyl transferase deficiency will commonly present as a newborn with jaundice, difficulty feeding, irritability, and hepatomegaly. Permanent damage to the brain and liver are inevitable without an early diagnosis. Classic cataract of galactosemia is an oil droplet located in the nucleus. Other causes of galactosemia include galactokinase deficiency. Diagnosis of galactosemia is demonstrated by increased levels of reducing substance in the urine. Treatment includes avoiding milk products and foods containing galactose.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|