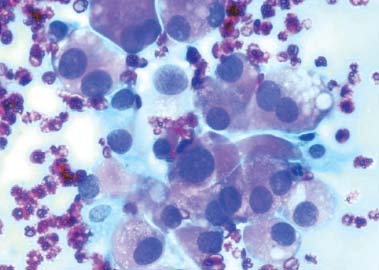
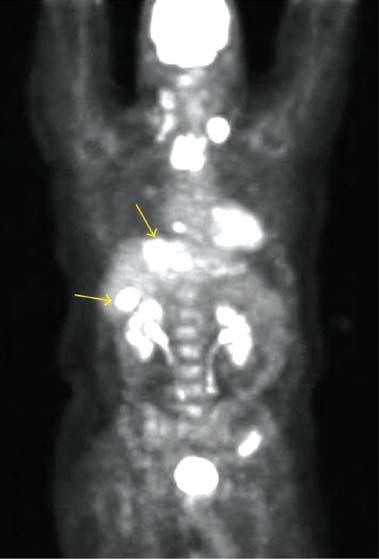
6 Core Messages • Medullary thyroid carcinoma (MTC) is a neuroendrocrine tumor of the calcitonin-producing thyroid parafollicular C cells and accounts for approximately 4% of thyroid cancers in the United States. • Approximately 78% of MTC is sporadic, with the rest inherited (familial) and usually part of a multiple endocrine neoplasia type 2 (MEN2) syndrome. There is no age-adjusted difference in survival between sporadic and MEN2A MTC, while the more aggressive MEN2B MTC has shown worse survival rates. • Diagnostic imaging, which can also examine for locoregional and distant spread, includes radiography, ultrasound or computed tomography (CT) of the neck; radiography or CT of the chest; CT of the upper abdomen; and nuclear medicine scintigraphy or positive emission tomography. • Confirmatory diagnosis involves pathologic examination of a fine-needle aspiration or surgical specimen. • Curative treatment always involves at least partial thyroidectomy, with total thyroidectomy usually recommended. Chemotherapy, radiotherapy, and immunoradiotherapy have a role in residual or recurrent disease, while radioiodine therapy is not used. • Biochemical follow-up involves trending serum calcitonin and carcinoembryonic antigen laboratory values. • Prognosis depends mostly on the tumor’s stage, with age being a controversial factor. Medullary thyroid carcinoma (MTC) is a neuroendocrine neoplasm of the thyroid parafollicular C cells and accounts for approximately 4% of thyroid cancers, or approximately 1000 cases per year, in the United States.1 The C cells are located in the upper two-thirds of the thyroid lobes and produce the polypeptide hormone calcitonin, which decreases serum calcium levels. While the thyroid epithelial cells are derived from the embryonic endoderm, the C cells develop from the neural crest, and therefore MTC shares clinical and histological features with other neuroendocrine tumors. MTC is unique among thyroid neoplasms for its inherited tumor syndromes; approximately 78% of MTCs are sporadic, while the rest appear to be inherited (familial), usually as part of the multiple endocrine neoplasia type 2 (MEN2) syndrome.2 The malignancy typically presents in the fifth or sixth decade of life for sporadic MTC and in the second or third decade for the inherited type.2 As mentioned earlier, approximately 22% of MTC are inherited (usually as part of MEN2), so MTC is traditionally classified as sporadic versus inherited. As first discovered in 1993, patients with inherited MTC have dominantly acting germline mutations in the RET proto-oncogene. The RET protein is a tyrosine kinase receptor that, in conjunction with the accessory molecule glial cell line-derived neurotrophic factor receptor-a, can transduce the glial cell line-derived neurotrophic factor signal via a pathway involving Ras and mitogen-activated protein kinase3,4; this pathway affects the differentiation, survival, and proliferation of a variety of neuroendocrine cell types. Inherited MTC is subclassified into the following categories: • MEN2A: MTC and pheochromocytoma + primary parathyroid hyperplasia. • MEN2B: MTC and pheochromocytoma + mucosal neuromas, intestinal ganglioneuromas, and a marfanoid habitus with a decreased upper/lower body ratio; this subtype has proven to be more aggressive than MEN2A with worse survival rates.5 • Familial medullary thyroid carcinoma (FMTC): MTC and no extrathyroidal manifestations. MEN2A also includes two minor variants: MEN2A with Hirschsprung disease (hypoplasia of the intestinal myenteric plexus) and MEN2A with cutaneous lichen amyloidosis. In addition, approximately 5% of the patients with FMTC have no detectable RET mutations and approximately 6% of the patients with sporadic MTC are believed to possess an unknown germline RET mutation.6–10 Although MTC can cause symptoms related to hypocalcemia from calcitonin overproduction (such as parasthesias, tetany, carpopedal spasms, petechiae, and hyperactive deep tendon reflexes), approximately 75 to 95% of the patients first present with a solitary thyroid nodule.2,11,12 Because the C cells are clustered in the upper portion of each thyroid lobe, most nodules are identified there. These nodules may interfere with or become more prominent during swallowing, and for locally advanced or metastatic disease, they may even cause hoarseness, dysphagia, or respiratory difficulty. Cervical lymphadenopathy may be appreciated, indicating locoregional spread. Patients who present with advanced disease may complain of weight loss, lethargy, bone pain, hemoptysis, or pneumonia symptoms (from aspiration-obstructive or postobstructive pneumonia), while their physical examination may demonstrate abdominal pain, jaundice (from liver metastases), or superior vena cava syndrome. Less commonly, patients with large tumor burdens may present with paraneoplastic syndromes, such as Cushing or carcinoid syndrome, with associated secretory diarrhea and flushing. Fine-needle aspiration or surgical biopsy with immunostaining for calcitonin is performed for patients presenting with suspicious nodules. MTC pathology reveals enlarged, spindle-shaped, pleomorphic tumor cells with eccentrically displaced nuclei (Fig. 6.1); there is no thyroid follicle development because the cells originate from the parafollicular C cells. The cytoplasm may also be slightly granular. However, the biopsy may reveal only C-cell hyperplasia (CCH), not a malignancy. Reactive CCH is caused by stimuli outside the C cell, such as hypercalcemia, hyperparathyroidism, chronic lymphocytic thyroiditis, or thyroid follicular tumors, and is usually not premalignant. By contrast, neoplastic CCH is caused by an intrinsic mutation and is a precursor to MTC. Figure 6.1 Fine-needle aspiration cytology reveals plasmacytoid cells with abundant cytoplasm and eccentric pleomorphic nuclei. Source: Mehdi G, Maheshwari V, Ansari HA, Sadaf L, Khan MA. FNAC diagnosis of medullary carcinoma thyroid: a report of three cases with review of literature. J Cytol 2010;27(2):66–68. Various extrathyroidal malignancies can metastasize to the thyroid, including renal cell, breast, melanoma, uterine, and lung carcinoma. If a tumor is suspected to be a metastasis, thyroglobulin staining as well as other markers should be administered to the biopsy specimen to determine the cells’ origin. Diagnostic imaging, which can also examine for locoregional and distant spread, includes radiography, ultrasound, or computed tomography (CT) of the neck; radiography or CT of the chest; CT of the upper abdomen; and nuclear medicine scintigraphy or positron emission tomography (PET). At presentation, spread to local lymph nodes in the neck is common, especially for inherited MTC. Other frequent sites of spread include mediastinal lymph nodes, liver, lung, and bone. An example of PET scan illustrating metastatic MTC is shown in Fig. 6.2. Spread to the skin or brain is also seen at presentation but is far less common. In addition to secreting excessive calcitonin, MTC also produces carcinoembryonic antigen (CEA); therefore, calcitonin and CEA levels are routinely measured to provide more information about the malignancy, its spread, and its activity. The biochemical preoperative analysis provides a baseline for comparison after the thyroidectomy to evaluate the effectiveness of the surgery. While hypercalcitoninemia is usually associated with MTC, this biomarker is not specific for the malignancy13; other conditions produce high levels of calcitonin, including hypercalcemia, hypergastrinemia, other neuroendocrine tumors, renal insufficiency, papillary and follicular thyroid carcinomas, and goiter.13 Indeed, only 10 to 40% of the patients with elevated levels of calcitonin and a thyroid nodule have MTC.14 However, very high levels of calcitonin (> 100 pg/mL) are essentially pathognomonic for MTC, with a risk approaching 100%, while the risk is approximately 25% for values 50 to 100 pg/mL, 8.3% for values 20 to 50 pg/mL, and with normal risk for values less than 8.5 pg/mL for men and less than 5.0 pg/mL for women.15 For patients diagnosed with MTC, it is important to determine whether the malignancy is part of an inherited syndrome. Patients should be asked about a family history of thyroid cancer, especially in first-degree relatives. In addition, a detailed personal and family history should include the presence of pheochromocytoma, hyperparathyroidism, mucosal neuromas, intestinal ganglioneuromas, or any of the other abnormalities associated with MEN2. A 24-hour urinary excretion of metanephrine, catecholamine, and cortisol should be measured to evaluate for pheochromocytoma. Some patients may elect to undergo genetic testing involving polymerase chain reaction amplification of the commonly mutated RET exons to determine whether they are at risk for other tumors as part of the MEN2 syndrome. This analysis of germline mutations in the RET proto-oncogene is the gold standard for determining this risk.16–18 Figure 6.2 Positron emission tomography scan showing extensive uptake in the neck, chest, and liver. Source: Gorospe EC, Badamas J. Acute liver failure secondary to metastatic medullary thyroid cancer. Case Reports in Hepatology 2011 doi:10.1155/2011/603757.
Medullary Thyroid Carcinoma II
Classification
Signs and Symptoms
Diagnosis and Multiple Endocrine Neoplasia Type 2
Medullary Thyroid Carcinoma II
Only gold members can continue reading. Log In or Register to continue

Full access? Get Clinical Tree


