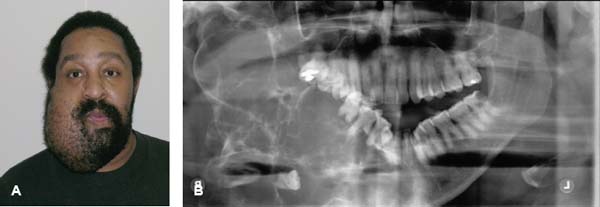
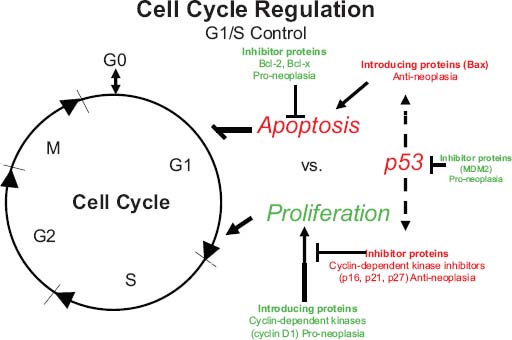
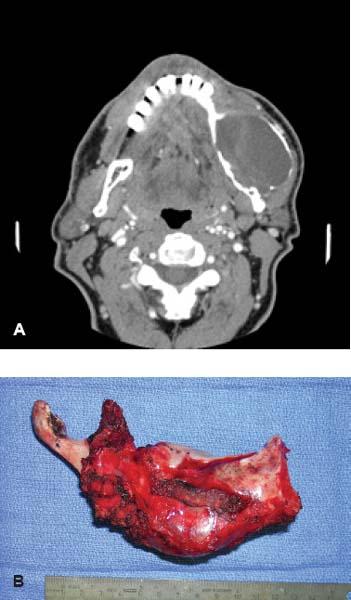
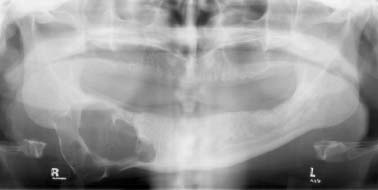
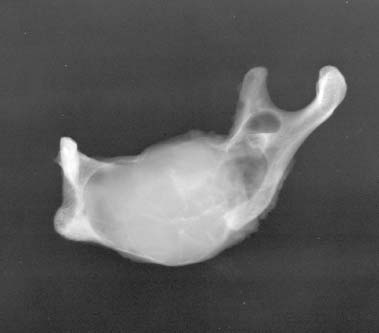
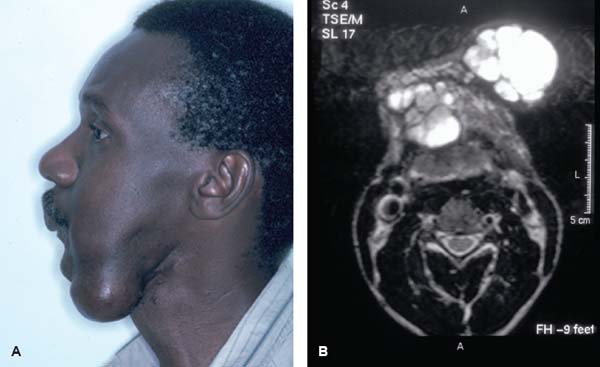
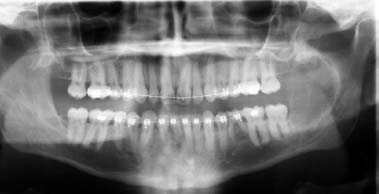
24 Core Messages • Although slow-growing, benign odontogenic tumors can be as destructive and life threatening as the more rapidly growing malignant tumors of the head and neck. • Like other head and neck neoplasms, odontogenic tumors exhibit growth patterns and doubling times that are a function of the interaction of cytokines and tumor suppressor genes related to proliferation versus apoptosis. • Langerhans cell histiocytosis represents a group of benign neoplastic processes comprising three morphologically similar lesions: eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease. • Fibro-osseous lesions frequently require the review of a radiograph of the pathologic process for proper diagnosis. The designation of a benign or malignant classification to a pathologic process of the jaws is based on specific histologic criteria. These criteria include the histologic presence or absence of necrosis and mitotic figures as well as a basic understanding of the specific entity under consideration. The term aggressive has most commonly been used to describe malignant neoplasms because of their ability to grow quickly and invade surrounding structures that may result in significant local growth, metastatic disease, and death of the patient. This notwithstanding, the jaws are the site of many locally aggressive benign neoplasms that may also result in significant hard and soft tissue destruction and deformation of the patient (Fig. 24.1), with resultant loss of function and possible airway obstruction. Aggressive benign processes of the jaws may be distinguished from their malignant counterparts by the lack of skin invasion, the lack of epineural infiltration, and the observed enigma of aggressive growth despite slow growth as may occur in many of these benign processes. In some cases, benign tumors of the jaws may be more aggressive, destructive, and deforming than some of their malignant counterparts, despite the observation that the benign tumors grow more slowly than malignant tumors. This concept is not only a paradox but also highly controversial, poorly understood, and often disputed. In addition, the slow growth of many aggressive benign neoplasms of the jaws has often resulted in subtherapeutic conservative surgical treatment with the thought that a recurrence will be diagnosed in an early time frame with the performance of more radical surgery. Benign neoplasms are dysmorphic tissue proliferations that have the capacity for persistent, autonomous growth. These tumors have the ability to progress unless completely removed. When completely removed, their reappearance is generally considered to not be possible, with a cure realized. When benign jaw tumors reappear following surgical extirpation, they are often incorrectly described as recurrent when, in fact, they really represent persistent disease because of earlier incomplete removal. In addition, the mere mention of a metastatic benign tumor of the jaws is seemingly a contradiction of terms, despite being occasionally diagnosed. Malignant neoplasms, by distinction, are best described as dysmorphic proliferations of tissues that have the capacity for autonomous growth, and metastasis, as well. The genetic alterations present in malignant tumors allow for a change in doubling times and for the development of metastatic disease after showing no previous capability for metastasis.1 The genetic alterations present in benign tumors are generally not susceptible to mutations, thereby conferring a relatively stable clinical course and prohibiting the development of metastatic disease. This genetic stability is what translates to relative consistency regarding fast versus slow growth, locally aggressive versus indolent infiltration of the surrounding tissues, and other clinical features of benign tumors. As mentioned earlier, a paradox exists with regard to the aggressive behavior of these benign processes and their rate of growth. Growth of all pathologic lesions occurs in the cell cycle as a function of cell proliferation versus apoptosis (Fig. 24.2). Neoplasia is understood, therefore, as a cell cycle disease.2 Alterations of genes that control the cell cycle are essential for the development of neoplastic disease. As such, neoplasia is characterized by a series of genetic alterations involving both oncogenes and tumor suppressor genes. Cell division comprises four phases including gap 1 (G1), DNA synthesis (S), gap 2 (G2), and mitosis (M). A major molecular event is the progression from the G1 to the S phase. Genetic alterations, if unrepaired in the G1 phase, may be carried into the S phase and perpetuated in subsequent cell divisions. This G1-S checkpoint is normally regulated by a well-coordinated and complex system of protein interactions whose balance and function are critical to normal cell division. Over production of inducing proteins or under production of inhibitor proteins may encourage the development of neoplastic disease. For example, p53, located on chromosome 17p13.1, is a tumor suppressor gene in its wild-type form. Normal p53 acts as a “molecular policeman” in its monitoring of the integrity of the genome. If DNA is damaged, wild-type p53 accumulates and inhibits replication to permit extra time for repair mechanisms to act. If repair fails, p53 may trigger cell suicide by apoptosis.3 As such, p53 serves as a negative regulator at the G1-S checkpoint. A mutation of p53 can be expected to allow cells to proceed into the S phase of the cell cycle before DNA can be repaired, thus encouraging the development of a tumor. Figure 24.1 A very large ameloblastoma of the mandible exhibiting clinical signs of severe facial deformation (A) and radiographic evidence of significant bone destruction (B). This tumor had been present for at least 20 years according to the patient. The future understanding of the involvement of p53 in tumor biology began in 1969 when Li et al reviewed medical records and death certificates of 648 childhood rhabdomyosarcoma patients and identified four families in which siblings or cousins had a childhood sarcoma.4 These four families also had striking histories of breast cancer and other neoplasms, suggesting a new familial cancer syndrome of diverse tumors. This syndrome took the name of Li–Fraumeni syndrome. Since the original description of the syndrome, systematic studies and anecdotal reports have confirmed its existence in various geographic and ethnic groups. The spectrum of cancers in this syndrome has been determined to include breast carcinomas, soft tissue sarcomas, brain tumors, osteosarcoma, leukemia, and adrenocortical carcinoma.5 Possible component tumors of this syndrome include melanoma, gonadal germ cell tumors, and carcinomas of the lung, pancreas, and prostate. These diverse tumor types in family members characteristically develop at unusually early ages, and multiple primary tumors are frequent. The molecular etiology of this syndrome is now known to be related to a germ line mutation of one p53 allele. Patients are therefore predisposed to develop malignant tumors as only one additional “hit” is required to inactivate the second, normal allele. Such individuals are said to have a 25-fold greater chance of developing a cancer by the of age 50 years compared with the general population.6 Once believed to be very unique, the understanding of p53 increased in 1997 with the discovery of another tumor suppressor gene called p73.7,8 Located on chromosome 1p36, this gene encodes a protein that bears many similarities to p53. It has a DNA-binding domain that resembles the corresponding region of p53, and similar to the latter it can cause cell cycle arrest as well as apoptosis under appropriate conditions.7,8 Figure 24.2 The cell cycle—a process of proliferation versus apoptosis (programmed cell death). A complex series of molecular events permits the proliferation of human tumors. Reprinted with permission from: Guild for Scientific Advancement in Oral and Maxillofacial Surgery. Carlson ER, August M, Ruggiero SL. Locally aggressive benign processes of the oral and maxillofacial region. Selected Readings in Oral and Maxillofacial Surgery 2004. As seen in Fig. 24.2, the expression of Bcl-2 and Bcl-x may also encourage neoplasia by inhibiting apoptosis, and MDM2 may directly inhibit p53. The Bcl-2 proto-oncogene was initially discovered at the break point of the t(14;18) chromosomal translocation in follicular lymphomas.9 Bcl-2 gene product protects tumor cells by blocking postmitotic differentiation from apoptosis, thus maintaining a stem cell pool. Bcl-x gene, a Bcl-2 homolog, encodes two proteins: a long form, Bcl-xL, and a short form, Bcl-xS.10 Bcl-xL protein, structurally and functionally similar to Bcl-2, has antiapoptotic activity, while Bcl-xS protein promotes apoptosis by inhibiting Bcl-2. Bax gene, an additional Bcl-2 homolog, encodes a protein that induces apoptosis. Bax protein regulates apoptosis by interacting with Bcl-2 or BclxL proteins. The MDM2 oncogene on chromosome 12q13–14 encodes a nuclear phosphoprotein that interacts with both mutant and wild-type p53.11 Its transcription is enhanced by wild-type p53 through the existence of a p53-binding site in an intronic sequence in the MDM2 gene. Both proteins regulate each other, forming an autoregulatory feedback loop that in turn regulates the transcriptional function of p53 protein and subsequent expression of the MDM2 gene. The MDM2 gene product can inhibit p53-mediated transactivation by masking the N-terminal acidic transactivation domain of p53 protein. High levels of MDM2 may inactivate the tumor suppressor activity of p53 by complexing to it. Therefore, deregulation of MDM2 function may be closely associated with tumorigenesis and/or tumor development. While the interaction of various cytokines with the cell cycle originally focused on malignant tumors, subsequent research identified that these cytokines and tumor suppressor genes are involved in molecular events related to benign tumor development, including many benign tumors of the jaws. Proliferating cell nuclear antigen is a cell cycle related antigen and has been used for the evaluation of the proliferation ability of many types of tumors and their recurrences. The detection of Ki-67 antigen is also a means to assess tumor cell proliferation.12 The monoclonal Ki-67 antibody was originally produced to a nuclear antigen in Hodgkin and Reed Sternberg cells, which was expressed in all proliferating cells during the G1, S, G2, and M phases of the cell cycle, but was absent in the G0 phase of the cell cycle.13 The observation of low rates of proliferating cell nuclear antigen and Ki-67 positivity in nuclei of some locally aggressive benign tumors of the jaws supports the observation of their slow growth. This observation would also support the perceived paradox of slow yet aggressive growth. Surgery for locally aggressive benign processes of the jaws is a function of the biologic behavior of the lesion. The principles of linear margins and anatomic barrier margins are important to consider. Linear margin principles refer to the inclusion of uninvolved soft and hard tissues surrounding the tumor specimen. Their inclusion occurs because of the understanding that tumors are not perfectly demarcated entities. Rather, tumors extend beyond their clinical and/or radiographic margins, such that inclusion of normal tissue assists in the likelihood of complete removal. Anatomic barriers are soft and hard tissues that surround a tumor and attempt to forestall its growth and infiltration of uninvolved tissues. The best known example is a capsule. Capsules surround some, but not all benign tumors. Unencapsulated benign neoplasms include the ameloblastoma and the pleomorphic adenoma. Other anatomic barriers include cortical bone, periosteum, muscle, mucosa, dermis, and skin. Locally aggressive benign tumors of the jaws often grow slowly such that violation of the skin does not occur (Fig. 24.1). Epidermal cells are thought to regenerate every 8 days, compared with a benign jaw tumor doubling time of several months or years. As such, the skin is able to maintain its integrity in the presence of the developing tumor and does not become violated by the tumor. True neoplasia is removed with attention to linear margins and anatomic barrier margins. The specific linear margin is a function of the histopathologic diagnosis of the neoplasm. Further, the removal of one uninvolved anatomic barrier margin with the tumor specimen seems to ensure complete removal (Fig. 24.3). An ameloblastoma that appears to be confined to the medullary component of the mandible, for example, may be able to be resected in a subperiosteal fashion. This notwithstanding, the surgeon may wish to proceed with a supraperiosteal dissection because of the fact that computed tomography (CT) scans may show intact cortical bone throughout, while the cortex may be perforated between CT cuts. The routine sacrifice of periosteum under such circumstances prevents entrance into tumor with inadvertent spilling. Odontogenic tumors represent a diverse group of pathologic entities that are of great interest to all surgeons of the head and neck. While a majority of these processes are centrally located neoplasms, some are believed to more accurately represent hamartomatous proliferations, and some may occur peripherally in soft tissue. True odontogenic neoplasms demonstrate varying inductive interactions between odontogenic epithelium and odontogenic ectomesenchyme. This ectomesenchyme was formerly referred to as mesenchyme as it was thought to be derived from the mesodermal layer of the embryo. It is now known and accepted that this tissue differentiates from the ectodermal layer in the cephalic portion of the embryo; hence, the designation ectomesenchyme. Figure 24.3 An ameloblastoma of the left mandible. The computed tomography scans show possible perforation of the lingual cortex of the mandible (A). As such, the anatomic barrier of periosteum and mylohyoid muscle was included on the medial aspect of the tumor specimen (B) so as to permit a clean margin in this area. Several of the aggressive odontogenic neoplasms represent prototypical examples of principles for benign tumor surgery of the jaws. These include the ameloblastoma, odontogenic myxoma, and the Pindborg tumor. They are true neoplasms that require attention to detail regarding bony linear margins and the surrounding anatomic barriers when performing extirpative tumor surgery. They also exemplify the observed paradox of aggressive local growth despite slow growth. The ameloblastoma is a benign tumor of the jaws and surrounding soft tissues that is characteristically locally aggressive. The literature and our experience indicate that these neoplasms are capable of significant destruction and deformation of facial structures, and occasionally death.14 The infiltration of surrounding soft and hard tissues of the face by the ameloblastoma is more outstanding than that accomplished by some malignant neoplasms of this anatomic area. In general terms, the aforementioned comments relate to the solid or multicystic variant of the ameloblastoma that requires a well-executed ablative surgery for cure. This notwithstanding, the unicystic ameloblastoma is, at times, also capable of significant destruction of the jaws. The unicystic ameloblastoma should not, therefore, be clinically underestimated and may require aggressive ablative surgery for cure. The peripheral ameloblastoma is, in contrast, innocuous and relatively indolent, thereby requiring relatively conservative surgery for cure. A review of 3677 cases of ameloblastoma found 2% to be peripheral, 6% to be unicystic, and the remaining 92% to be solid or multicystic.15 Studies from North American authors find the ameloblastoma to comprise approximately 10% of all odontogenic tumors.16,17 The solid or multicystic ameloblastoma is the most common variant of ameloblastoma15 and the most widely discussed.18 It is also the variant whose treatment is perhaps the most controversial. As Regezi et al2 and Gold18 point out that this tumor was identified well more than a century ago, with either Cassock or Broca being credited with the first scientific report of the ameloblastoma in 1827 and 1868, respectively.19 The solid or multicystic ameloblastoma is primarily a tumor of adults, occurring predominantly in the fourth and fifth decades, with an average age of occurrence of the early 30s19. While this variant of ameloblastoma is rare in children, studies document their existence.20,21 These studies also point to the preponderance of the unicystic variant when a diagnosis of ameloblastoma in children is made. This variant of the ameloblastoma may occur throughout the maxilla or mandible, but has a predilection for the posterior mandible. In a study of 98 ameloblastomas by Mehlisch et al, 91 (93%) were located in the body or ramus of the mandible while 7% occurred in the symphysis.22 Ueno et al reviewed their findings with 104 ameloblastomas, of which 97 occurred in the mandible.23 Of the mandibular ameloblastomas, 94 cases (97%) occurred in the molar region, 60 cases (62%) occurred in the ramus, while 28 cases (29%) occurred in the symphysis of the mandible. The maxilla is an infrequent site for the solid or multicystic variant of the ameloblastoma.24–26 When this tumor involves the maxilla, approximately 90% occur in the posterior maxilla.19 Figure 24.4 An ameloblastoma present in a 27-year-old male who presented with facial swelling. The panoramic radiograph shows a multilocular radiolucency. The solid or multicystic ameloblastoma is most commonly asymptomatic, but may occasionally produce a painless mass. Pain, tooth mobility, and trismus are less common findings.23 Radiographically, the solid or multicystic ameloblastoma most commonly appears as a multilocular radiolucency (Fig. 24.4). Because these processes are slow growing, the radiographic margins are usually well-defined and sclerotic. Treatment of the solid or multicystic ameloblastoma has been a source of contention in the oral and maxillofacial surgery literature for decades. While most would agree that this neoplasm is aggressive and deserves aggressive surgical management from the outset, there are several authors who advocate conservative treatment initially and reserve radical surgery for recurrences.27–29 As pointed out previously, labeling tumors that reappear following conservative surgery as recurrences probably represents a misnomer, with persistent disease more accurately describing the clinical outcome. Conservative surgical management of this variant of the ameloblastoma has historically included enucleation and curettage, while aggressive or radical surgery has involved resection. Those who recommended curettage have relied on the belief that ameloblastoma invades cancellous bone but not cortical bone.30 As pointed out by Carlson,1 however, the cortical bone represents a competent anatomic barrier that may not be violated by a very small ameloblastoma. Larger tumors, however, show obvious clinical, radiographic, and histologic evidence of cortical bone invasion by the ameloblastoma. It should be clear that the advancing front of the tumor is beyond the radiographic or clinical margin, thereby requiring the inclusion of a linear margin of bone in the tumor surgery. Other problems associated with curettage of this neoplasm include the violation of one of the first premises of tumor surgery that is to not spill tumor. An enucleation and curettage surgery, by definition, enters the tumor and likely predisposes the patient to persistent disease. It is our contention, therefore, that conservative measures have no role to play in the surgical management of the solid or multicystic ameloblastoma. Resection of the ameloblastoma with negative soft and hard tissue margins should be expected to result in cure of the patient. In his review of patients with ameloblastoma treated in a variety of ways, Mehlisch et al revealed a “recurrence” rate of 90% for those patients treated with curettage, while infrequent “recurrence” for those patients treated with resection.22 Sehdev et al reviewed 92 patients with ameloblastoma and noted curettage to be followed by “local recurrence” in 90% of mandibular ameloblastoma and 100% of maxillary ameloblastomas.26 Equally worrisome was the finding that subsequent resection was able to control 80% of mandibular ameloblastomas and resection of “recurrent” maxillary ameloblastomas was ineffective in controlling the tumor. In the final analysis, terms such as radical or conservative should probably not be used to describe the treatment of the ameloblastoma. A scientific understanding of this tumor is that it is a slow-growing, aggressive, benign neoplasm that is best controlled and cured with a resection with approximately 1.0 cm bony linear margins.31 The surgeon may wish to verify the bony linear margin with an intraoperative specimen radiograph so as to provide security that a sufficient bone margin was sacrificed while the patient is still generally anesthetized (Fig. 24.5). Close bone margins noted on the specimen radiograph may be addressed with additional resection of bone. Cure of the patient should be realized if tumor is noted histologically to be well-contained within included anatomic barriers on the specimen. Under such circumstances, patients can be subsequently reconstructed and fully rehabilitated dentally. Attempts to control this tumor with more conservative measures compromise these objectives. Salvage with radiation therapy has been described for the management of ameloblastoma.32,33 Once thought to be radioresistant, the ameloblastoma has been proved to respond to radiation therapy in limited series. The value of such therapy is realized in those cases where a full surgical excision would be technically difficult because of bulk and local invasion or where other medical factors, including age, would make radical surgery inappropriate. In the review by Atkinson et al,32 2 of the 10 patients underwent an attempt at surgical control of the tumors, yet with incomplete excision. As such, postoperative radiation therapy was offered, and the patients showed no evidence of disease at 30 and 60 months postoperatively. Of the remaining 8 patients, 6 showed no evidence of disease at a range of 1 to 10 years following the delivery of radiation therapy without surgical intervention. In 2 of these 8 patients, a residual mass was noted long after the conclusion of radiation therapy. We believe that radiation therapy should not be necessary as part of the therapy for ameloblastoma when primary surgery is executed properly. Radiation therapy may be considered for use, however, in the postoperative management of relatively nonresectable tumors that have previously been subtherapeutically managed with enucleation and curettage surgeries (Fig. 24.6). Pathogenetic mechanisms of the solid or multicystic ameloblastoma include the expression of Bcl-210,34 and MDM2.11 In their review of 25 surgical specimens of ameloblastoma by Mitsuyasu et al,34 12 of which were unicystic ameloblastomas, and 13 of which were solid or multicystic tumors, all were noted to express Bcl-2 protein, mainly in the outer layer of tumor cells. The stellate reticulum and squamoid cells were negative. In addition to inhibiting apoptosis, the Bcl-2 protein was felt to play a role in maintaining the stem-cell population in the peripheral layers of the tumor nests from which proliferating cells are recruited. Kumamoto and Ooya10 studied the expression of Bcl-2 and Bax proteins in various types of ameloblastoma. The findings were the presence of Bcl-2 protein expression in ameloblastomas in the peripheral cells neighboring the basement membranes. Bcl-x protein was distributed similarly to Bcl-2 protein, but was expressed more extensively than Bcl-2 protein. Reactivity for bax protein was quite low in ameloblastomas. Carvalhais et al11 examined the expression of MDM2 in 13 ameloblastomas and a variety of other odontogenic lesions. These ameloblastomas showed higher MDM2 expression than radicular cysts, but lower than the two groups of odontogenic keratocysts. This notwithstanding, the presence of MDM2 gene expression by ameloblastomas supports their pathogenesis. Figure 24.5 A specimen radiograph of an ameloblastoma resection showing at least 1 cm linear bony margins in the proximal and distal aspects of the resection. The specimen radiograph provides an intraoperative assessment of the adequacy of the specimen’s bone margins. Figure 24.6 The clinical appearance (A) and magnetic resonance imaging (B) of a patient following two enucleation and curettage surgeries for ameloblastoma of the mandible. At this time, he displays soft tissue persistence of his tumor in the facial skin. In addition, substantial floor of mouth and pharyngeal extension of the tumor was noted. The patient underwent a skin sacrificing wide excision of this persistent tumor followed by the administration of postoperative radiation therapy. One very important question that has surfaced in the literature is whether or not the ameloblastoma is malignant. Discussions by Willis35 and Carr and Halperin36 exemplify this debate. Willis stated that attempts to distinguish between benign and malignant ameloblastoma are futile in that they are all malignant in that they are locally invasive and prone to recur. Carr and Halperin stated that malignant and benign are poor terms when applied generally to the ameloblastoma. Furthermore, they stated that the use of one term versus the other seems secondary to an understanding of how the tumor may behave and to an approach to treatment based upon this understanding. Gold justified his belief that all ameloblastomas are malignant in drawing an analogy between basal cell carcinoma of skin and the ameloblastoma.37 He indicated that the basal cell carcinoma is slow growing, infiltrative, capable of great destruction of soft tissue and bone, recurrent when not completely eradicated, capable of invading vital structures, seldom metastatic but capable of metastasis, presenting several histologic patterns, and arising from the epithelial skin surface and skin adnexa. He rationalized that if one substitutes oral epithelium for skin surface and dental lamina/enamel organ for skin adnexa, one would be describing the ameloblastoma. Despite his argument, Gold lamented that the long-held traditional belief that the ameloblastoma is benign will be difficult to reverse. Finally, as agreed by most surgeons and pathologists, the ameloblastoma is distinctly aggressive, infiltrative, and unpredictable in its behavior. In 1977, Robinson and Martinez reviewed 20 patients presenting with unilocular cystic lesions whose clinical, radiographic, and growth features were those of dentigerous or primordial cysts.38 On the basis of morphology, the epithelial islands and portions of the lining epithelium seen in all of the 20 cases were indistinguishable from ameloblastic epithelium, the characteristics of which have been described by Vickers and Gorlin.39 This feature of the unicystic ameloblastoma has been disputed by Gardner and Corio40 who indicate that the basal cells are not remarkable and do not fulfill the criteria of Vickers and Gorlin for ameloblastoma. A review of the literature would suggest that the term unicystic developed from the observation that most of these lesions were, in fact, unilocular radiographically. Regezi et al41 has recommended the term cystic ameloblastoma because of the identification of an occasional multilocular lesion. In any event, three well-accepted histologic subtypes of this variant of the ameloblastoma have been noted, including the luminal, intraluminal, and mural subtypes. While the unicystic ameloblastoma in general can be treated conservatively with a high rate of cure, the mural subtype should be discussed separately, owing to its different biologic behavior.42 With an inherently more aggressive behavior, it is considered one of the locally aggressive benign tumors of the jaws and ought to be treated similarly to the solid or multicystic ameloblastoma.43 The average age of occurrence for the unicystic ameloblastoma is the mid-twenties. In general, this is younger than that of the solid or multicystic ameloblastoma. In Robinson and Martinez’s series of 20 patients, the mean age was 27.7 years. This finding has been confirmed by other authors.40,41,44 The site of predilection for this variant of the ameloblastoma is the mandible, with the molar/ramus region being most commonly affected (Fig. 24.7). This tumor is frequently associated with an impacted tooth. Eversole found six radiographic patterns in his review of 31 cases of unicystic ameloblastoma.45 In all six patterns, the lesions were radiolucent and well-defined. Three radiographic patterns were observed in cases where these lesions were associated with impacted third molars, and three radiographic patterns were seen in cases that were not associated with an impacted tooth. Four of the six patterns were distinctly unilocular. The author stressed that any large unilocular or multilocular radiolucency in a child, teenager, or young adult should develop suspicion for the presence of a unicystic ameloblastoma. Historically, the literature regarding the unicystic ameloblastoma has found a much lower rate of “recurrence” following curettage compared with that of the solid or multicystic ameloblastoma. The series by Robinson and Martinez38 showed a 25% “recurrence” rate following curettage, and Gardner and Corio41 reported a “recurrence” rate of 10.7% following curettage. In general, the luminal and intraluminal variants of the unicystic ameloblastoma are readily cured with an enucleation and curettage surgery (Fig. 24.8). The mural variant of the unicystic ameloblastoma is, because of its anatomic location, less likely to be cured with this type of surgery. Most of the studies discussing the unicystic ameloblastoma do not separate the mural variant from the more favorable luminal and intraluminal variants. As such, we believe that the inclusion of the mural variant in discussion of cure rates negatively impacts the cure rates of the unicystic ameloblastoma as a whole. In other words, the luminal and intraluminal variants of the unicystic ameloblastoma are highly curable lesions when performing an enucleation and curettage surgery. The mural variant of the unicystic ameloblastoma, however, is truly a locally aggressive benign neoplasm of the jaws whose biologic behavior parallels that of the solid or multicystic ameloblastoma. As such, the mural subtype of the unicystic ameloblastoma should be treated with resection34,43 (Fig. 24.9). As has been pointed out, a definitive diagnosis of the mural subtype of the unicystic ameloblastoma may occasionally be made following an enucleation and curettage surgery, under which circumstances the surgeon may wish to adopt close follow-up rather than committing the patient to a return to the operating room for resection. Serial panoramic radiographs should be obtained at regular intervals so as to promptly diagnose the presence of persistent disease with subsequent resection. If the diagnosis of a mural subtype of the unicystic ameloblastoma is made based on incisional biopsy, we recommend primary aggressive management with resection. Figure 24.7 A unicystic ameloblastoma of the left mandible presenting as a unilocular radiolucency.
Benign Aggressive Jaw Tumors
Odontogenic Tumors
Ameloblastoma
Solid or Multicystic Ameloblastoma
Clinical and Radiographic Features
Treatment and Prognosis
Unicystic Ameloblastoma
Clinical and Radiographic Features
Treatment and Prognosis
Benign Aggressive Jaw Tumors
Only gold members can continue reading. Log In or Register to continue

Full access? Get Clinical Tree


