48
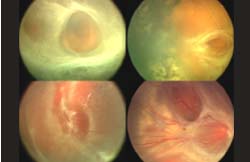
Macular Disease Secondary to Peripheral Retinal Vasculopathy
Maximiliano Gordon  Hugo Quiroz-Mercado
Hugo Quiroz-Mercado  R.V. Paul Chan
R.V. Paul Chan
Peripheral retinal vascular changes are common findings associated with both local and systemic disease in the pediatric population. These changes can be helpful in differentiating between various disease states resulting from primary ocular conditions well known to the ophthalmologist or systemic disease that requires a careful clinical history and systemic workup in order to determine the etiology.
Peripheral retinal vascular changes may result in significant visual impairment, especially if the macula is involved. Patient age, extension of vascular changes, and duration of disease all contribute to the prognosis. Of major concern, however, is the presence of peripheral retinal ischemia with secondary retinal neovascularization (1). If this is to occur, appropriate management of the patient can preserve and/or improve macular function. Laser treatment is the treatment of choice and should be directed toward the peripheral retina, but at times direct intervention to the macular area may need be considered. Herein we describe the most common diseases associated with peripheral retinal vascular changes, and we focus mainly on those conditions that require laser photocoagulation, cryotherapy, or surgical intervention. Tables 48-1 and 48-2 show a classification for differential diagnosis (1–3). Pathologies not included in this classification are the inflammatory diseases associated with vasculitis like Eales’ disease, Behcet’s disease, sarcoidosis, systemic lupus erythematosus, multiple sclerosis, pars planitis, and other less common problems that are described in other chapters.
PATHOPHYSIOLOGY
Peripheral retinal abnormalities such as retinal neovascularization, vascular tortuosity, ectasis, vascular shunts, and aneurysms may damage the inner blood-retina barrier. Damage to the retinal vascular endothelium may involve primary arteries, veins, or capillaries, or any combination of the three. Also, the endothelial alteration may be focal or widespread. Fluorescein angiography has demonstrated that the entire capillary bed may be affected in some cases, whereas in others, the changes may only be limited to capillaries in the midneuroretina, with the inner retinal vessels remaining normal.
TABLE 48-1 HEREDITARY OR CONGENITAL VASCULAR DISEASES (1–3)
Sickle cell retinopathy
Retinitis pigmentosa
Angiomatosis retinae (von Hippel’s disease)
Congenital retinal telangiectasis (Leber’s military aneurysms,
Coats’ syndrome)
Congenital retinal macrovessels and arteriovenous
communications
Incontinentia pigmenti
Retinal cavernous hemangioma
Inherited retinal venous beading
Small vessels hyalinosis
TABLE 48-2 ACQUIRED VASCULAR DISEASES (1–3)
Retinal capillary obstruction/loss
Retinopathy of prematurity
Hyperviscosity syndromes
Diabetes mellitus
Radiation retinopathy
Longstanding retinal detachment
Retinoschisis
Toxemia of pregnancy
Cocaine abuse
Choroidal melanoma and hemangioma
Decreased ocular blood supply
Ocular ischemic syndrome
Carotid cavernous fistula
Encircling sclera buckling operation
Decreased retinal blood supply
Large retinal vessels obstruction
Retinal embolization
Retinal venous occlusive disease
Following surgical retinectomy
The extracellular space of the retina generally is considered to be relatively small compared with other tissues except for the brain. The outer plexiform layer is the primary interstitial space in the retina. If the retina becomes edematous, it is in this layer that fluid accumulates in the outer plexiform layer. The macular contains only four layers of the retina: the internal limiting membrane, the outer plexiform layer, the outer nuclear layer, and the rods and cones. The absence of Muller cells in the foveal region is also a contributing factor (4). No intermediate layers exist between the internal limiting membrane and the outer plexiform layer in the fovea, which in the macula is oblique (outer plexiform layer of Henle). This is an important factor in understanding the stellate appearance of the cystoid edema in the macula as opposed to the honeycomb appearance of cystoid edema outside the macula (3, 5).
According to the severity of the damage in the blood-
inner retina barrier, Gass distinguished three categories of macular edema: mild, moderate, and severe (3). If the decompensation is mild, small molecules and proteins escape into the extracellular space, and clear serous exudate may be confined to the inner retinal layers. It is not visible biomicroscopically. In fluorescein angiography, diffuse mild staining of the inner retina is observed. If capillary damage is moderate, deeper plexus of capillaries are affected. Serous fluid accumulates within the inner nuclear and outer plexiform layers. The biomicroscopic picture of cystoid macular edema then can be observed. Swelling of the retina and loss of the foveal depression is caused by the development of large central cysts. On fluorescein angiography, molecules diffuse out of the capillaries, producing a stellate pattern. If endothelial damage is severe, large proteins and lipids escape into the extracellular compartment, and the exudate may be cloudy. The
extravascular protein is transported across the pigment epithelium, choroid, and sclera. Around the outer margin of capillary leakage, the fluid is reabsorbed and small ions that are components of the blood leave behind the lipoprotein in the outer plexiform layer that tend to aggregate frequently in a circinate pattern.
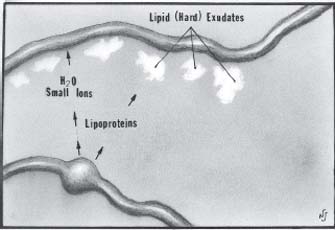
Figure 48-1. Schematic diagram of leakage from retinal aneurysm. Small ions and water are reabsorbed. The large lipoprotein molecules are too large to enter the healthy capillary wall, and they deposit along these vessels, frequently in a circinate pattern. (Adapted from Ferris FL, Patz A. Macular edema, a complication of diabetic retinopathy. Suv Ophthalmol 1984;28(suppl):452–561, with permission.)
After resolution of the capillary leakage or following photocoagulation, macrophages remove the lipid exudates (3, 6) (Fig. 48-1).
In diseases with severe vascular abnormality involving the peripheral retina, chronic gravitation of the subretinal lipid to the macula and inferior periphery may cause widespread deposits of subretinal and outer retinal exudate remote from the vascular abnormality (3, 7). Massive lipid residue may cause permanent damage to the retina and pigment epithelium, as well as choroidal neovascularization (8). Early treatment of vascular lesions may prevent permanent visual lost and resolution of lipid deposits in the macula (9).
Vitreous changes and membranes formation secondary to peripheral vascular diseases may produce macular ectopia, macular traction with or without tractional retinal detachment, rhegmatogenous retinal detachment, and epimacular membrane (10, 11)
RETINOPATHY OF PREMATURITY
Retinopathy of prematurity (ROP) is a potentially blinding eye disease (12). It has been suggested that therapeutic oxygen, although important, has been overemphasized as a cause of ROP under contemporary neonatal care practices. Other factors related to very low birth weight are probably quite important, especially in view of current nursery monitoring of oxygen. Birth weight is inversely related to risk of ROP and is at least as good an indicator as is gestational age. With current nursery practices, ROP is truly a disorder of the “smallest and sickest” infants (13).
Classification
The International Classification of ROP (ICROP) and the Cryotherapy for ROP (CRYO-ROP) trials have had a profound impact on the way in which we manage ROP (12).
The CRYO-ROP study provided a classification system for ROP which categorized the disease into zones and stages. Zone I uses the optic nerve as the center of a circle, and the radius is defined as two times the distance between the foveola and the optic nerve. Zone II uses as a radius the distance between the nasal ora serrata in the horizontal meridian and the center of the optic nerve. All of the remaining retina is zone III (14–16).
CRYO-ROP also defined plus disease, a descriptive term for six clock hours of dilated and tortuous vessels of the posterior pole. In addition, the anterior segment in plus disease often shows dilated iris vessels (14–16).
Preplus disease has been defined as vascular abnormalities of the posterior pole that are insufficient for the diagnosis of plus disease but shows more arterial tortuosity and more venous dilatation than normal. Over time, these vessels may dilate and become more tortuous, progressing to plus disease (13).
There are five stages of ROP. Stage 1 signifies a narrow white line present at the junction of vascular and avascular retina. Stage 2 is a ridge of activity with thickening of this line. Stage 3 involves the growth of extraretinal fibrovascular proliferation at the ridge. Stage 4 is a partial retinal detachment and is subclassified as 4-A, with the macula attached, and 4-B, with the macula detached. Stage 5 implies a total detachment of the vascularized retina (14–16)(Fig. 48-3). Aggressive, posterior ROP is a rapidly progressive, severe form of ROP seen in very low birth weight infants. The fundus appearance is characterized by prominent plus disease and flat neovascularization. It is currently seen most commonly in zone I or posterior zone II (12, 14).
Treatment
The CRYO-ROP study determined that treating threshold disease defined as five contiguous or eight cumulative clock hours of stage 3 with plus disease was beneficial over observation. The Early Treatment for ROP (ETROP) study indicated beneficial results for any eye with zone I stage 3, zone I with plus disease, zone II stage 2 or 3 ROP with plus disease. Early treatment showed better visual and structural outcomes in premature infants over a 10-year period (12).
Currently, laser photocoagulation is the treatment of choice for treatment requiring ROP. As better visualization of the retina is possible and technology has advanced, cryotherapy for ROP has fallen out of favor (17).
Anti-vascular endothelial growth factor (anti-VEGF) agents (e.g., bevacizumab) have recently been advocated for the treatment of ROP. Quiroz-Mercado, Martinez-Castellanos et al. reported the use of bevacizumab (Avastin), injected intravitreally for ROP. Thirteen patients (18 eyes) were included in the study. Patients were separated in three different groups: group I included patients with stage IVa or IVb ROP who had no response to conventional treatment (cryotherapy or laser); group II included patients with threshold ROP who could not receive treatment secondary to poor visualization of the retina; and group III included patients with high-risk prethreshold or threshold ROP. Regression of neovascularization occurred in 17 eyes. One patient with stage IVa ROP had spontaneous retinal reattachment after a single intravitreal injection of bevacizumab. There were no serious ocular or systemic adverse events reported (18).
Although retinal ablation is effective in most cases of treatment requiring ROP, a significant number of these eyes progress to retinal detachment (stages 4A, 4B, and 5) (12). The natural history arm of the CRYO-ROP study showed that a child with 8 sector 4A ROP at their due date (40 weeks PMA) has a high risk of going on to an unfavorable outcome or total retinal detachment (stage 5) (12, 19, 20).
Surgical options for ROP advancing to retinal detachment include scleral buckle (SB), vitrectomy with lensectomy, lens sparing vitrectomy (LSV), and open sky vitrectomy. Although scleral buckling for stage 4B and 5 ROP may provide an anatomic outcome superior to the natural history of the disease, this approach does not provide visual results as rewarding as one would hope because of induced anisometropia and amblyopia. Nor does scleral buckling deal directly with vitreous traction (19).
There are numerous advantages to LSV over SB for tractional stage 4A ROP retinal detachments. First, SB has an anatomic success rate on the order of only 70%. Second, placement of a SB requires an additional procedure to divide the encircling element so that the eye may continue to grow. Third, scleral buckling could produce an induced mean anisometropia of −9.5 diopters, with residual myopia on the order of −5 diopters, even after the encircling element is divided. Fourth, visual acuity results for stage 4A detachments repaired with scleral buckling surgery techniques have been very discouraging.
Although visual acuity has not yet been measured accurately in children with LSV, the potential for very good visual acuity should be high based on the central, steady, and maintained fixation behavior noted to date (19).
Capone and Trese designed a study to assess the efficacy of LSV in tractional 4A ROP retinal detachments in reducing progression to stage 4B or 5 ROP. The study included forty eyes (31 patients) with stage 4A ROP at 38 to 42 weeks postconceptional age. Pars plicata vitrectomy was performed on all patients. An infusion light pipe, vitreous cutter, and membrane peeler cutter (MPC) scissors were used in the surgical technique. At the last follow-up examination, 36 of 40 eyes showed complete retinal reattachment with central steady and maintained fixation. Four eyes progressed to 4B retinal detachments, and in three of those four eyes the retinas were reattached after repeat vitreous surgery. One eye progressed to stage 5 ROP. The 90% anatomic success rate of LSV for 4A ROP reported in the current series is far superior with regard to both anatomic outcome and visual prognosis. Also, it has been suggested that the ideal timing for vitreoretinal intervention is when the vascular activity (dilation and tortuosity) has abated and detachment has just begun (19).
In another series, Sears and Sonnie compared the anatomic outcomes of LSV with those of combined LSV and SB in surgical repair of ROP stage 4 retinal detachment. Twenty-one eyes of 15 patients with stage 4 ROP detachment were included. An SB was placed externally using either a 240 or 41 band after 360-degree conjunctival peritomy and isolation of the four rectus muscles. The SB was secured to the eye wall with 5-0 nylon sutures as close to the ridge as possible and was fastened with a Watzke sleeve. Drainage of subretinal fluid was not performed in any eye. All SBs were removed by six months of age. LSV was performed using two sclerotomies posterior to the iris root (1.5 mm from the limbus) through pars plicata at 9:30 and 2:30. An end irrigating Capone light pick was used in all cases in conjunction with a pediatric wide-angle viewing system and a 23-gauge pediatric MPC. Of the patients in whom treatment failed, two were in the LSV with SB group (2/12; 16%) and one was in the LSV alone group (1/9; 11%). Overall, the study results suggest that SB adds little to the success or failure of LSV and therefore is an unnecessary adjunct for stage 4 (A and B) (21).
Gonzales, Boshra, and Schwartz used 25 gauge pars plicata vitrectomy for stage 4 and 5 ROP. Fifteen eyes of 12 infants were included. Three-port pars plicata vitrectomy using 25-gauge instrumentation was performed. Conjunctival dissection was performed in all cases and sclerotomies were made 0.5 to 1.0 mm posterior to the limbus through the pars plicata. Several vectors of traction must be addressed including those extending from the ridge to the lens, from the ridge to the anterior vitreous base, and from the ridge to the optic nerve. Eleven of 15 (73%) eyes had documented retinal reattachment after one or more surgeries at the last follow-up. Complications included vitreous hemorrhage and postoperative cataract. But they concluded that 25-gauge vitrectomy is a safe and effective treatment approach for tractional retinal detachments in stage 4 and 5 ROP (22). Retinal photocoagulation or cryotherapy may be effective for stabilizing aggressive posterior ROP; however, it very frequently cannot stop the progression to retinal detachment (23).
In an attempt to address this issue, Azuma et al. studied the efficacy of early vitrectomy for aggressive posterior ROP to stop progression of retinal detachment. Twenty-two eyes (15 patients) with aggressive posterior ROP underwent vitrectomy with or without lens sparing, because retinal photocoagulation failed to stop progression of fibrovascular proliferation, despite being performed early, densely, and with early retreatment. Six eyes (100%) in which an LSV was performed developed a large tractional retinal detachment. In contrast, the retinas were completely reattached in 16 eyes (100%) in which vitrectomy with lensectomy was performed, nine eyes (56%) had foveal configuration, and 14 eyes (88%) had steady fixation. These results indicate the great benefit of early surgery for aggressive posterior ROP, in comparison to the poor visual outcomes after vitreous surgery for Stage 5 ROP (23).
FAMILIAL EXUDATIVE VITREORETINOPATHY
In 1969, Criswick and Schepens reported six children with peripheral retinal abnormalities resembling ROP but distinguished by their familial occurrence, and no history of prematurity or supplemental oxygen after birth. Systemic associations were absent (24). They named the disease familial exudative vitreoretinopathy (FEVR).
The clinical findings include heterotopia of the fovea with temporal traction, organized vitreous membranes, peripheral neovascularization with abrupt termination of the temporal retinal vasculature, retinal exudates, retinal folds and tractional retinal detachment. Anterior chamber structures are uninvolved. Nonetheless, the end stages of severely affected eyes may display chronic retinal detachment with cataract, band keratopathy, and glaucoma (24).
FEVR is always bilateral and usually symmetric. Some infants have family members with similar findings, indicating that this is an autosomal dominant condition, with nearly 100% penetrance. Like other autosomal dominant conditions, expression is variable, with some members having only mild macular dragging or small areas of peripheral avascularity of the retina, demonstrable only by fluorescein angiography. X-linked recessive trait, with high penetrance, and variable expressively, and sporadic cases have also been reported (24).
The pathogenesis of FEVR appears to be a consequence of disturbed development of the retinal vasculature in the last months of gestation, with a failure of the peripheral retina to vascularize. Although the ensuing changes bear a similarity to the pathobiology of ROP, they follow a different time course and natural history. A notable difference is the tendency of ROP to progress to cicatricial stages or to abort and vascularize the periphery, whereas the avascular zone in FEVR remains a permanent feature throughout life (24).
A classification system, based on the ophthalmoscopic findings, has been proposed by Pendergast and Trese. The disease is classified into five stages (Table 48-3) (25).
Management
As mentioned previously, it is important to identify FEVR by careful examination of blood relatives. In most asymptomatic cases, observation and frequent follow-up are all that is required. In children with strabismus, recognition, again, is important. Treatment with cryotherapy or laser ablation of the neovascular areas, and scleral buckling and vitrectomy procedures for tractional detachments, may be required (26).
TABLE 48-3 CLINICAL CLASSIFICATION OF FAMILIAL VITREORETINOPATHY

Pendergast and Trese reported the results of surgical management of FEVR. Fifty-two eyes of 26 patients with FEVR were studied. A total of 40 eyes were treated. Seven eyes required no treatment and five eyes had inoperable retinal detachments. Fifteen eyes were treated with peripheral laser ablation initially and 25 eyes presenting with retinal detachments required vitreoretinal surgery. Of the 15 eyes treated initially with laser, eight eyes required no further treatment, whereas seven eyes progressed to retinal detachment requiring vitreoretinal surgery. A total of 32 eyes (including seven previously lasered eyes) underwent vitreoretinal surgery. Patients with retinal detachment or macula heterotopia were treated with vitrectomy, scleral buckling, or both. Vitrectomy was performed using a two-port system with an infusion light pipe. Whenever possible, a lens-sparing approach was used (Fig. 48-2). In eyes in which the vitreoretinal membranes extended anteriorly and prevented safe entry through the pars plicata, entrance wounds were made at the limbus through the iris root and a lensectomy was performed. Twenty-nine of these 32 eyes had at least 6 months of follow-up. At the last follow-up visit, the macula was attached completely in 18 eyes (62.1%) (25).
In a retrospective study, Ikeda et al. examined the anatomic features and surgical indications of FEVR complicated with rhegmatogenous or tractional retinal detachment. Twenty-eight eyes of 25 patients who had either clinically suspected or fully diagnosed FEVR were included in this study. Of these,
25 had rhegmatogenous retinal detachment, two had tractional retinal detachment, and one had tractional retinal detachment plus vitreous hemorrhage. The vitreoretinal adhesions were so strong in the peripheral avascular area that iatrogenic retinal breaks easily occurred in 22 of 28 eyes. the surgeons attempted to remove the posterior vitreous membrane as far into the equator as possible and then combined this procedure with broad scleral buckling from the equator to the vitreous base against the residual vitreoretinal traction (27).
Joshi et al. reviewed six eyes (four ROP, two FEVR) with diffuse contraction of the posterior hyaloid resulting in retinal detachment. They observed a diffuse proliferation along the posterior hyaloids that required extensive lamellar dissection at times aided by autologous plasmin enzyme. A diffuse, taut posterior hyaloid resulted in a marked tractional component to retinal detachment. Bimanual dissection of the contracted hyaloid from the retinal surface was essential in relieving tractional force on the retina. The study concluded that hyaloid contraction aids in the surgical planning and repair of these complex eyes (28).
INCONTINENTIA PIGMENTI
Incontinentia pigmenti (IP) is an X-linked dominant disorder with characteristic cutaneous (Fig. 48-4), dental, skeletal, central nervous system, and ocular manifestations; it is lethal in most male embryos. The precise pathogenesis of the clinical manifestations of IP is unknown. Clinical suspicion of the diagnosis is usually a result of the characteristic rash that develops at or shortly after birth and is confirmed by skin biopsy, showing intraepithelial vesicles filled with eosinophils. Systemic manifestations often become evident within the first 4 months of life and are seen in almost 80% of patients (29). Neurologic deficits occur in one-third of patients and can manifest as hydrocephalus, microcephaly, and mental retardation (30). Ocular involvement occurs in 35% of persons with IP; findings include strabismus, conjunctival pigmentation, cataracts, corneal epithelial and stromal keratitis, iris hypoplasia, optic atrophy, and, most important, vitreoretinal abnormalities (29).
The vitreoretinal manifestations are persistence of fetal vasculature, foveal atrophy, paramacular vascular dilatations and aneurysms, retinal pigment epithelium mottling, retinal pigment epithelium hypopigmentation, peripheral retinal avascular zones and vascular tortuosity, arteriovenous anastomoses, preretinal neovascularization at the junction of vascular and avascular retina, vitreous hemorrhage, and tractional
retinal detachment (Fig. 48-4). Retinal tears and rhegmatogenous retinal detachments most likely occur as a result of peripheral vitreoretinal traction at the junction of avascular and vascular retina (29).
It is difficult to establish prophylactic or therapeutic guidelines for vitreoretinal involvement in IP, because the relatively small number of cases has precluded careful study of the natural history of the disease. Cases with vascular changes without neovascularization or exudation probably do not need treatment (31), whereas cases with progression of neovascularization and evidence of exudation can be treated with photocoagulation (32). Patients with vitreous hemorrhage or tractional retinal detachment should be treated with vitrectomy.
DIFFERENTIAL DIAGNOSIS OF PEDIATRIC VASCULAR DISEASE
ROP, FEVR, and IP may resemble the less common bilateral pathology of Norrie’s disease or the unilateral pathology of persistent fetal vasculature (PFV).
NORRIE’S DISEASE
Norrie’s disease is an X-linked recessive disorder that can present with retinal folds or vascularized retrolental masses simulating retinoblastoma (“pseudoglioma”). The principal features are bilateral severe retinal dysplasia and optic nerve hypoplasia, associated in some cases with hearing loss and progressive mental deterioration (33, 34).
PERSISTENT FETAL VASCULATURE
This condition is one of the most common congenital abnormalities affecting the eye (35). The syndrome is usually an isolated monocular finding in an otherwise healthy child and has a wide spectrum of anatomic and pathology features (36). As Goldberg points out, the term “persistent hyperplasic primary vitreous” is a misnomer in common usage, because it fails to include persistence of other components of the fetal intraocular vasculature. PFV is secondary to the persistence of the vascular component that is freely anastomotic in fetal life; hence they may persist together in varied degrees of severity (35). It shows lack of involution of prenatal vessels after
birth (36).
Usually, this is a unilateral disorder characterized by abnormal regression of the primary vitreous and fetal vessels encasing the posterior surface of the lens (36). Unilateral cases of PFV are sporadic, while bilateral cases may be associated with X-linked Norrie disease. Associated sensorineural deafness and mental retardation in males may be absent, and genetic testing can be useful in separating sporadic PFV from Norrie disease (37).
PFV is subclassified into three types: type 1—anterior PHPV (retrolental fibrovascular membrane, elongated ciliary processes, cataract, microphthalmia); type 2—posterior PFV (vitreous membrane and stalk, retinal fold, traction retinal detachment, hypoplastic optic nerve and macula, microphthalmia); and, most common, type 3—a combination of anterior and posterior PFV. The purely posterior form occurs in only 10 to 20% of cases. Persistence of the hyaloid artery, Bergmeister papilla, nonattachment of the retina, retinal folds and macular hypoplasia are other features of the posterior type. In the pure form of posterior PFV, anterior structures are not involved, although microcornea and an immature filtration angle are often present (38).
Nystagmus occurs in bilateral cases. With age, the anterior chamber shallows, resulting in glaucoma or corneal decompensation in some eyes (36).
The differential diagnosis of PFV includes intraocular nematodes such as Toxocara sp and cysticercosis, occult intraocular foreign body, endophthalmitis, and X-linked retinoschisis.
Management
Without treatment, severely affected eyes invariably progress to phthisis bulbi (36, 38). The visual prognosis in patients with PFV is often poor, especially in the posterior form of the disease. Surgical treatment may not be effective in patients with severe microphthalmia or advanced posterior PFV, such as marked foveal hypoplasia, dysplasia, rhegmatogenous retinal detachment (RRD), and retinal or optic nerve abnormalities. These patients may need only to be observed (39).
Two surgical methods have been proposed to manage PFV associated with retrolenticular membranes: an anterior transpupillary approach and a posterior pars plana/plicata approach (40).
Payse et al. believe that surgical avoidance of the ciliary body reduces the risk of retinal tears, retinal detachment, and inadvertent retinectomy. In this study, three clear corneal ab externo paracentesis incisions were created approximately 0.25 mm central to the limbus using a 19-gauge microsurgical vitreoretinal blade. An anterior chamber maintainer attached to the infusion solution was placed in the inferotemporal incision. An anterior capsulotomy was then performed using a vitrector (vitrectorhexis). The easily accessible soft cortical lenticular material was then aspirated. Then, an attempt was made to incise the retrolenticular material with the vitrector. If the above measures were unsuccessful, intraocular vitreoretinal scissors were inserted. Multiple radial cuts created wedge-shaped membrane segments small enough to be removed with a vitrector. (40, 41).
In a retrospective study by Alexandrakis et al., patients with PFV were divided into two groups: one surgical and the other nonsurgical, and the final best postoperative visual acuity, prognostic, ocular clinical features, and surgical complications were compared. In the surgical group, 30 patients were included. Two patients had clinical and echographic findings consistent with anterior PFV, two had strictly posterior PFV, and the remaining 26 had components of both anterior and posterior PFV. Indications for surgery included media opacity (e.g., cataract), vitreoretinal traction, and retinal detachment. The two patients with strictly posterior PFV underwent a pars plana vitrectomy, and the remaining 28 patients underwent a primary pars plana lensectomy and vitrectomy procedure. Of the latter 28 patients, 12 also had a membrane peeling procedure and two underwent a scleral buckling procedure at the time of vitrectomy for a TRD. Two patients had a posterior chamber intraocular lens placed at the time of lensectomy. In the group of 12 patients who were observed, the decision was based on the presence of mild PFV (2 patients), foveal or optic nerve hypoplasia or both (6 patients), marked microphthalmia (3 patients), and inoperable RRD (1 patient). Approximately 50% of patients undergoing surgery for PFV will achieve useful vision. Visual acuity outcomes in patients with PFV are correlated with the nature and extent of ocular risk factors. Some patients may not be candidates for surgery because of either minimal changes or advanced disease that limit the potential of visual improvement (39).
COATS’ DISEASE
First described by George Coats in 1908, Coats’ disease is a disease characterized by retinal telangiectasia and exudation of unknown etiology (42), which exhibits a wide spectrum of clinical findings (43). In addition to telangiectasias, there may be capillary nonperfusion, aneurysm formation, exudation both within and beneath the retina, and massive lipid deposition (44) (Fig. 48-4). It is predominantly a nonhereditary disease of childhood that is diagnosed within the first decade of life. It affects males more than females and is unilateral in about 90% of patients. Vision loss, strabismus and leukocoria may occur and it is critical that Coats’ disease be differentiated from retinoblastoma (44). Less severe manifestations can be observed in adults (42). If the disease is not treated, it can lead to total retinal detachment and secondary glaucoma, occasionally requiring enucleation (45).
Chronic retinal detachment is associated with exacerbation of retinal vascular abnormalities, as a result of impaired oxygenation of the outer retina and secondary expression of increased VEGF or a similar growth factor that may cause the microvascular changes seen in Coats’ disease. For some patients, anti-VEGF therapy may be a potential treatment candidate (44).
Based on their observations, Shields et al. classified Coats’ disease in five stages (Table 48-4) (45).
TABLE 48-4 CLASSIFICATION OF COATS’ DISEASE

The term Leber’s miliary aneurysm refers to the adult form, with intraretinal exudation commonly affecting the macula with yellowish circinate exudation, cystoids macular edema, subretinal detachment, and less commonly exudative retinal detachment (46). Less severe presentations, commonly observed in 40-year-old men, have retinal telangiectasis confined to a small segment of juxtafoveolar area. These patients constitute type I juxtafoveolar telangiectasis (47).
Treatment
The main goal of treatment should be to eradicate the telangiectasias in order to facilitate resolution of exudation and salvage as much vision as possible. Without treatment, the natural progression of the disease may lead to complications such as total bullous retinal detachment, neovascular glaucoma, and phthisis bulbi (42).
Patients with stage 1 disease (telangiectasia only) can be managed by either periodic observation or laser photocoagulation. However, encountering a patient with stage 1 disease is uncommon in clinical practice (45). Patients with stage 2 disease (telangiectasia and exudation) are generally best managed by laser photocoagulation or cryotherapy, depending on the extent of the disease (45). Patients with stage 3A disease (subtotal retinal detachment) can generally be managed by photocoagulation or cryotherapy. Because laser photocoagulation is less effective in areas of retinal detachment, cryotherapy is often preferable in such cases (45). Stage 3B (total retinal detachment) can be managed with cryotherapy if the detachment is shallow, but may require an attempt at surgical reattachment if the detachment is bullous and immediately posterior to the lens. Surgical repair of total and bullous retinal detachment is justified only to prevent the development of neovascular glaucoma, even though the visual outcome is expected to be poor (45). Stage 4 disease (total retinal detachment and glaucoma), is often best managed with enucleation to relieve the severe ocular pain (48). Patients with stage 5 disease generally have a blind, but nonpainful eye, and require no aggressive treatment (45).
Observation is recommended in two situations: in eyes with stage 1 and 2A disease with mild telangiectasia and little
exudation, particularly in patients older than 15 years, because in these cases there is less likelihood of progression of exudation and retinal detachment. However, treatment should be considered if progression is documented. Also, in some eyes with stages 3B and 5 disease, where the blind eye is comfortable, but there is no hope for useful vision, observation is an option (45).
Laser photocoagulation is most successful when there are telangiectasias without retinal detachment (stage 2). Cryotherapy can also be used in such cases. It is important to wait at least 3 months before considering additional laser photocoagulation (45).
Cryotherapy is the treatment of choice when there are peripheral telangiectasias associated with extensive exudation or subtotal retinal detachment (stage 3A), even in cases of relatively high retinal detachment (stage 3B) (45).
In a retrospective study by Shienbaum and Tasman the average follow-up period for the 13 patients was 12.4 years. Four out of the twelve treated patients (33%) had recurrences, and three of the four had multiple recurrences (42).
New treatment modalities have been tested using anti-VEGF agents. Quiroz-Mercado et al have been using intravitreal bevacizumab (Avastin) injections in patients with Coats’ disease as an adjuvant treatment before cryotherapy. Four eyes of four patients with Coats’ disease were included in a study. Partial resolution of subretinal fluid was observed (49).
PERIPHERAL RETINAL ANGIOMA
Peripheral retinal angiomas as well as the entity described as Leber’s military aneurysm, represent a challenge in diagnosis and treatment for the vitreoretinal surgeon. Peripheral retinal hemangiomas have been described as sporadic lesions or as part of the von Hippel-Lindau syndrome (50, 51). The term “retinal angioma” to most ophthalmologists is synonymous with the lesions seen in von Hippel-Lindau syndrome. Welch (52) and McDonald (53) categorized other angioma-like lesions as pseudoangiomas. They emphasized that if the lesion has all the clinical characteristics of the retinal capillary hamartoma, it represents a case of von Hippel-Lindau syndrome until proven otherwise.
Shields et al. (54), in a review of 103 vasoproliferative retinal tumors of the ocular fundus (acquired retinal hemangioma), created a comprehensive classification. Shields found that 74% of the tumors were idiopathic, and 26% were secondary to preexisting ocular disease (pars planitis, retinitis pigmentosa, or toxoplasmic retinitis). Most of the idiopathic tumors developed within 6 mm of the ora serrata. Associated vitreoretinal exudation, secondary exudative retinal detachment, vitreous cells, vitreous hemorrhage, preretinal macular fibrosis, and macular edema (52).
Peripheral retinal capillary hemangiomas have been well described as part of the von Hippel-Lindau syndrome, associated with hemangiomas in the cerebellum, medulla, or spinal cord; cysts of the pancreas, kidney, adrenal gland; and hypernephroma and pheochromocytoma (55).
Vision may be lost as result of cystoid macular edema, subretinal exudates, serous retinal detachment, vitreous hemorrhage, rhegmatogenous retinal detachment, or macular traction created by epiretinal membranes or tractional retinal detachment (53, 56) (Fig. 48-7).
Treatment
Cryotherapy and laser photocoagulation have been shown to be effective in treating peripheral angiomas (57). Cryotherapy and laser treatment to peripheral retinal angiomas may induce “spontaneous peeling” of epiretinal membranes (53, 58). Most epiretinal membranes that spontaneously peel away from the macula are in eyes with partial posterior detachment, in which complete posterior vitreous detachment later develops. Presumably, eyes that show spontaneous membrane peeling after cryotherapy or laser photocoagulation for peripheral angiomas do so because the treatment results in angioma permeability changes that enhance vitreous liquefaction and complete detachment.
McDonald et al. (53) reported the use of vitrectomy in eyes with macular traction associated with peripheral angiomas, ranging from macular puckering to frank tractional detachment of the macula associated with partial posterior vitreous detachment, peripheral tractional retinal detachment, and extensive epiretinal membrane formation. These changes may arise after cryotherapy, laser treatment, or spontaneously. Vitreous surgery has a good chance of improving vision in these cases; treatment of the hemangioma, before or during vitrectomy, usually results in tumor regression (53). Adequate treatment of hemangiomas can be accomplished by endolaser treatment, cryotherapy, or the use of indirect ophthalmoscopic laser photocoagulation.
Another treatment option that has been suggested is the use of intravitreal bevacizumab. Ustariz-Gonzalez, Quiroz-
Mercado, et al. treated two patients with intravitreal injection of bevacizumab resulting in improvement of visual acuity in both cases (−1.40 logMAR to −0.60 logMAR, and −1.40 logMAR to −0.80 logMAR). Vitreous opacities seen by ophthalmoscopy and color photos decreased in both eyes and flourescein angiograohy (FA) showed less leakage after the treatment. At 3 months follow up, no further changes were observed (59).
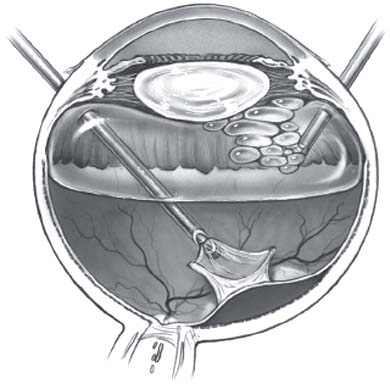
Figure 48-2. Lens-sparing vitrectomy in infants. Two steps of the technique are shown: vitrectomy-membranectomy and fluid-gas exchange performed by infusing air through the irrigating light pipe. (Adapted from Maguire AM, Trese MT. Lens-sparing vitreoretinal surgery in infants. Arch Ophthalmol 1992;110:284–286, with permission.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


