Ocular adnexal lymphoproliferative disorders (OALDs) comprise 10% of all extranodal disease and may involve any part of the orbital soft tissue, as well as the conjunctiva, eyelids, or lacrimal sac.1 Intraocular and optic nerve lymphomas are commonly associated with CNS disease and are not discussed in this chapter.
The benign end of the spectrum includes reactive lymphoid hyperplasia and IgG4-related ophthalmic disease.2 These lesions are slow-growing, solitary masses and are clinically similar to low-grade lymphomas. They are defined pathologically as having well-defined growth centers, including various mature cell types and polyclonal immunologic markers (Fig. 15.2). These are described in more detail in Chapters 11 and 17.

Ocular adnexal lymphomas (OALs) arise from monoclonal expansion of malignant B or T lymphocytes and are the most common orbital neoplasms, representing 55% of cases in adults. T and natural killer (NK) cell lymphomas are very rare; over 90% of orbital lymphomas are primary, B-cell, non-Hodgkin lymphomas (NHLs) (Fig. 15.3).

OAL may be a primary process, arising within ocular adnexal structures, but it may also occur secondarily from primary lesions elsewhere in the body. When there is a history of prior lymphoma elsewhere, followed by the development of a similar ocular adnexal lesion, the orbital process is obviously secondary. When there is simultaneous presentation of ocular adnexal disease and systemic disease, this potentially could represent a primary orbital process with secondary systemic involvement or a secondary involvement of the orbit from a primary more aggressive lesion elsewhere. Less commonly, the ocular adnexa may be involved by direct extension from primary lesions in adjacent sinuses.
Epidemiology
Lymphoma can be broadly divided into NHL (70%) and Hodgkin disease (30%).3
Hodgkin lymphoma is made up of clonal Hodgkin and Reed-Sternberg cells with an accompanying mixed cellular infiltrate. These tumors arise from B cells that have lost their B-cell phenotype.4 Orbital involvement is very rare.
In the United States, 72,000 new cases of NHL arise annually and cause 20,000 deaths each year.5 The annual incidence for mature B-cell neoplasms, which form the largest subgroup of lymphoma, ranges from only 1.2 in 100,000 in China to 20 in 100,000 in the United States, Europe, and Australia.5 The overall incidence of lymphoma has been increasing annually by 3% to 4% for many decades,6,7 but the rate of extranodal disease has been increasing at a greater rate.8,9 Within the extranodal subgroup, OALD has shown the greatest increase in incidence at a rate of up to 6.3% per year.10–12
Historical Background
OALD has confounded clinicians and pathologists for decades. Limitations in early pathology led to discrepancies between clinical course and pathologic appearance in 20% to 50% of cases.13–15 This mismatch applied to both morphologically benign histologic specimens (evaluated with hematoxylin and eosin stain) that followed an aggressive course, as well as lesions that appeared aggressive on histology but remained localized clinically.16,17
With the discovery of separate B- and T-lymphocyte subsets in the 1970s,18–20 and with the insights gained through electron microscopy,21 pathologists became aware that lymphoma comprised distinct entities. With immunophenotyping and the use of the cluster of differentiation (CD) nomenclature, these entities became more apparent.22,23 CD20, originally dubbed “B1,” was the first CD discovered; it is a pan-B-cell marker.
The application of molecular genetics to pathologic specimens has finally given pathologists the ability to define the genetic aberrations that underlie these lesions precisely. 24–26
Extranodal lymphomas of mucosa-associated lymphoid tissue (MALT) were described elsewhere in the body almost contemporaneously with those of the orbit, but had probably been described in the orbit previously.21,27,28 Since the early descriptions of MALT lymphoma, the importance of this entity in the ocular adnexae, where it is the most common subtype of lymphoma, is being appreciated to a greater extent.29–32
Historical classification schemes did not include OALD and also omitted extranodal lesions. The current iteration of the World Health Organization (WHO) modification of the Revised European American Lymphoma Classification (REAL) recognizes both extranodal disease and marginal zone lymphomas and is designed to accommodate new entities as further diagnostic progress elucidates additional subtypes.33,34
Pathogenesis
Normal Lymphocyte Development
Understanding lymphomagenesis requires an appreciation of the normal lymphocyte development and its role in the immune system.35 Lymphocytes need to respond to myriad antigens and develop an infinite range of different clones that can interact with each of them. Lymphomas generally represent malignant, clonal proliferations of lymphocytes. Many of the pathways and processes involved in the normal immune response are altered, allowing lymphoma development. These include genetic changes (which range from chromosomal translocations to more subtle somatic mutations and alterations of copy number), influence of the tumor microenvironment, and chronic antigen stimulation.36,37 The changes act on the B-cell receptor (BCR), P13K/AKT/mTOR, NF-kB, epigenetic regulatory, and cell cycle pathways.38
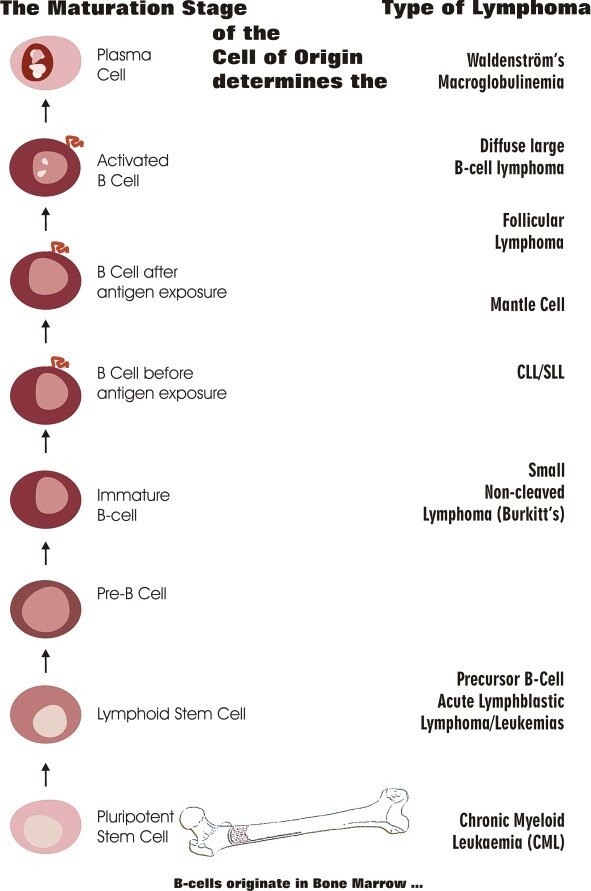
The WHO lymphoma classification subtypes largely correspond to clonal proliferations of cells arrested at specific stages of lymphocyte development34 (Fig. 15.4).
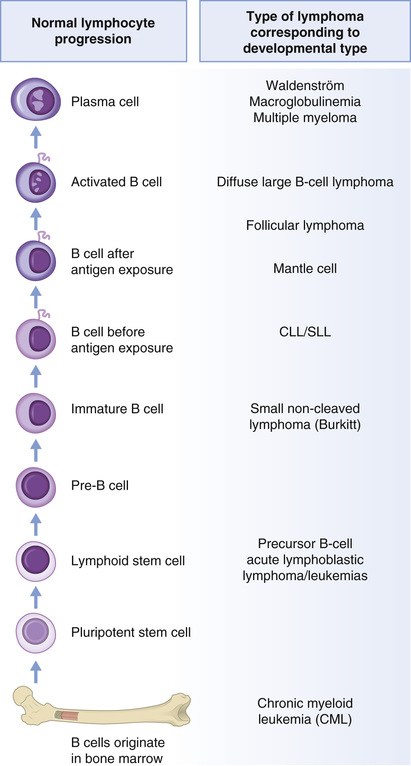
Normal lymphocyte development begins in the marrow with precursor B lymphoblasts, which undergo immunoglobulin variable, diversity, and joining recombination (VDJ recombination) to become surface immunoglobulin positive naive B cells, providing a diverse BCR repertoire (Fig. 15.5).38,39 These recirculating naive B cells are found in blood, primary lymphoid follicles, and follicle mantle zones. Exposure to antigen leads to transformation to blast cells, which then migrate into the dark zone of the germinal center of the primary follicle, where they are known as centroblasts, and continue to divide rapidly. Many of these early germinal center changes depend on the transcriptional repressor gene, B-cell lymphoma 6 (BCL6 gene).40 Here, the immunoglobulin heavy (IGH) and light (IGL) chain loci are subjected to additional genetic change by activation-induced cytidine deaminase (AID) somatic hypermutation (SHM), affording the B-cell receptor with improved antigen affinity. These centroblasts migrate to the light zone of the germinal center, are further challenged by T cells and follicular dendritic cells (FDCs), and undergo AID-mediated class switch recombination (CSR) to establish different immunoglobulin isotypes. The majority of these centrocytes are low affinity, and 90% are eliminated by apoptosis. BCL6 downregulation normally occurs now and is important for further lymphocyte development. About 10% remain, interact with surface molecules, and differentiate into memory B cells or plasma cells. The memory B cells are found in the marginal zone of the lymph follicle, whereas plasma cells home to marrow41 (Fig. 15.5).

Corresponding to these stages of development, Burkitt lymphoma derives from dark zone centroblasts, diffuse large B-cell lymphomas from germinal center light zone centroblasts and follicular lymphomas (FLs) from germinal center light zone centrocytes. Extranodal marginal zone lymphoma and lymphoplasmacytic lymphoma arise from memory B cells, whereas mantle cell lymphoma arises from mature naive B cells, found in the mantle region of the lymph node (Figs. 15.4 and 15.5).
A range of genetic (chromosomal translocations, deletions, and mutations) and epigenetic changes, as well as environmental (chronic antigen stimulation) and microenvironmental (tissue factors) changes, occur during the different phases of lymphocyte development, eventually establishing a clone of malignant cells.
Cytogenetic Influences
The acquisition of genetic aberrations in lymphocytes causes clonal cell proliferation and suppression of apoptotic mechanisms, immune suppression, and altered cell signaling functions that result in tumor initiation, promotion, and growth. When altered, the delicate balance between oncogenes and tumor suppressor genes leads toward lymphoid malignancy.
Certain cytogenetic abnormalities are characteristically seen in different lymphoma types. The first abnormality to be detected was the 8;14 translocation seen in Burkitt lymphoma, using standard karyotypic methods. Since that time, advances in molecular genetics have allowed for refined diagnosis of OALDs. Assessment with conventional cytogenetics, with cell culture or Southern blot methods, have been superseded by polymerase chain reaction (PCR) and fluorescent in situ hybridization (FISH) techniques, employed routinely in most pathology laboratories. Comparative genome hybridization (CGH) and other sophisticated research laboratory techniques permit analysis of DNA sequence copy number to detect loss or gain across the genome.42,43 More recently, high-resolution single nucleotide polymorphism array (SNP-A) karyotyping has been used to study chromosomal and genetic abnormalities of OALD in detail.44
The most common OALD is the extranodal marginal zone lymphoma (EMZL). These lymphomas show a range of cytogenetic abnormalities which vary from those seen in MALT lesions elsewhere in the body, such as the lung, gut, or skin. These include: t(11;18)(q21;q21) of the API2 and MALT1 genes (occurs in 0%–10% ocular adnexal EMZL), t(14;18)(q32;q21) of the IGH and MALT1 genes (occurs in 7%–11% ocular adnexal EMZL), t(1;14)(p22;q32) of the Bcl-10 and IGH genes (not reported to occur in ocular adnexal EMZL), and t(3;14) (p14;q32) of the FOXP1 and IGH genes (not reported to occur in ocular adnexal EMZL).36 These different abnormalities result in activation of the transcription factor NF-κB, which upregulates various proliferation genes in B cells. Other abnormalities seen include trisomy 3 (occurs in 40%–60% ocular adnexal MALT lymphoma) and trisomy18 (occurs in 14%–50% ocular adnexal MALT lymphoma).45 SNP-A has recently confirmed the most frequent copy number gain was trisomy 3 (31%), followed by trisomy 18 (17%), 6p, and 21q (14%), whereas copy number losses were seen in 6q and 9 p (7%).44 This technique also showed that although copy number variation occurred in roughly 70% of OALD, they were not seen in benign lymphoproliferative disorders.
The incidence of these cytogenetic abnormalities varies greatly with MALT lymphomas derived from different tissues, for example, gastric, lung, skin, and ocular adnexae.46 Interestingly, given the low percentage of ocular adnexal EMZL showing the translocations common in other MZL, other genetic, epigenetic, and microenvironmental factors are likely influencing extranodal marginal zone lymphomagenesis.
Follicular lymphoma (FL) develops from centrocytes and centroblasts of the germinal centers that fail to undergo apoptosis because BCL2 expression is preserved as a result of the initial t(14;18) chromosomal rearrangement.41 Additional genetic alterations occur, leading to FL, which may have a better or worse prognosis, depending on which secondary alterations take place.47
We have already learned that BCL6 plays an important role in germinal center formation and subsequent lymphocyte development. Failure to downregulate BCL6 after affinity maturation may be lymphomagenic.40,48 BCL6 is necessary for survival of human diffuse large B-cell lymphoma (DLBCL) cells. DLBCL commonly shows alterations of the BCL-6 gene at the 3q27 locus, but other complex karyotypes may be seen.49 These different abnormalities may explain the morphologically and immunohistochemically different centroblastic and immunoblastic subtypes of DLBCL.49 There are at least three distinct entities grouped together under the DLBCL banner based on distinct chromosomal imbalances. These are germinal center B-cell-like (best prognosis), activated B-cell-like (intermediate to poor prognosis), and a poor prognosis non GCB-like non-ABC-like subgroup.50
Mantle cell lymphoma (MCL), an aggressive lymphoma, develops from a combination of dysregulation of cell proliferation and survival pathways with a high level of chromosome instability. The genetic hallmark of MCL is the t(11;14)(q13;q32) translocation that juxtaposes CCND1, at chromosome 11q13, to the Ig heavy chain gene at chromosome 14q32.51 CCND1 is a proto-oncogene, which encodes cyclin D1, resulting in cyclin D1 overexpression. This translocation occurs in bone marrow in an early B cell at the pre-B stage of differentiation when the cell is initiating the immunoglobulin gene rearrangement with the recombination of the V(D)J segments. The cell of origin is a mature B cell found in the mantle region of normal lymphoid follicles. Although the initial translocation occurs in immature B cells in bone marrow, the oncogenic advantage is realized only when additional genetic aberrations occur as the cell matures into a naive pre–germinal center B cell.51,52 Diagnosis of this small cell lymphoma can be confirmed by immunohistochemical staining for cyclin D1 and with FISH techniques.53
T-cell malignancies comprise two main groups: precursor T-cell lymphoblastic neoplasms, derived from maturing thymocytes, and peripheral T-cell lymphomas (PTCLs), apparently arising from mature postthymic T cells. Physiologic T-cell development is regulated by numerous oncogenes and oncogenic pathways, suggesting a balance between normal differentiation and malignant transformation.54 Recent studies suggest that premalignant cells arise early in hematopoietic differentiation as a result of TET2 and DNMT3A mutations and further genetic insults in phenotypically mature T cells, including G17V RHOA mutations that lead to tumor development in a defined multistage process. This comprises a three-signal process with recurrent genetic modifications in antigen (“signal 1”), co-stimulatory (“signal 2”), or cytokine receptors (“signal 3”) and the tyrosine kinases and other signaling proteins they activate.55
For all of these lymphomas, the clinical behavior of the tumor usually reflects the behavior of the normal cell counterpart. This corresponds to the lymphocyte stage at which the abnormal cell has accumulated sufficient genetic abnormalities to proliferate without control, or avoid programmed cell death, constituting malignancy. Malignant cell clones that have low turnover produce indolent lymphomas such as EMZL and FL, whereas cell stages that are more active give rise to more aggressive lesions such as mantle cell or DLBCL. DLBCL has a number of subtypes derived from different genetic influences on the light zone centroblast.
A percentage of the low-grade lymphomas will undergo transformation to a higher grade lesion. FL transforms at roughly 2% to 3% per year into a higher grade lesion, with a molecular profile that closely resembles DLBCL. Four to 10% of EMZL can also transform into DLBCL (with a so-called triple hit rearrangement of c-MYC, BCL6, and BCL2) or more rarely into Hodgkin lymphoma. Risk factors include age greater than 60 years and elevated lactate dehydrogenase (LDH). Interestingly, these patients frequently have other nonlymphoid malignancies.
As mentioned earlier, the most common OAL is the EMZL. The orbit has no lymph nodes or true lymphatic drainage system but does have a well-established MALT system extending from the lacrimal gland to conjunctival tissues and the lacrimal drainage apparatus. This can be broken down into the “conjunctival-associated lymphoid tissue” and “lacrimal drainage–associated lymphoid tissue” (CALT, LDALT) with an overall designation of “eye-associated lymphoid tissue” (EALT).56 Lymphoid follicles from these tissues participate in the normal immune response to antigens with production of antibodies and effector plasma cells. Although most OAL is EMZL derived, presumably arising from these tissues, lymphocytes destined to reside in the EALT system pass through the normal lymphocyte development cycle, and this may explain why other primary B-cell lymphomas are seen in the ocular adnexal region. Lymphomas originating in ocular adnexal tissues can have systemic lymphoid involvement of bone marrow and other tissues. Conversely, systemic lymphomas may involve adnexal tissue secondarily.
Marginal zone–derived B cells home to their specific extranodal sites (e.g., cells destined for the gut migrate to the gut and not the ocular adnexa, and vice versa). Nodal B cells migrate to specific lymph nodes or secondary lymphoid organs such as the tonsils or thymus. There is site specificity for the homing of normal post–germinal center B lymphocytes, orchestrated by adhesion molecules, cytokines, chemokine and their receptors, sphingosine-1-phosphate receptors (SIPR), and integrins.57,58 These molecules play important roles in tracking between blood, secondary lymphoid organs, and extranodal sites such as the ocular adnexa. A recent pivotal study showed that such ocular adnexal lymphomas (regardless of histologic subtype) expressed a profile of molecules, suggesting a dynamic process of trafficking involving not only tissue retention but also egress via S1PR3 and homing back to extranodal sites via CXCR4/CXCL12 and α4.59 α4β1, expressed strongly in OAL, is essential for migration across venular endothelium toward sites of inflammation. This implies that anatomic location in B-cell NHL is orchestrated by variable expression of adhesion and motility molecules, and may help explain why OALD may arise at a site of chronic inflammation (Fig. 15.6), is frequently bilateral, and can spread to other predominately extranodal sites.

Chronic Antigen Stimulation
Chronic antigen stimulation and infectious agents have important influence in the pathogenesis of lymphomas. Chronic low-grade infection and inflammation may induce and promote carcinogenesis, altering DNA and providing a carcinogenic environment bathed in cytokines and growth factors.60 Ocular adnexal EMZL often develops in a setting of chronic inflammation61 and has been shown to be associated with Chlamydia psittaci, Helicobacter pylori, hepatitis C virus, and other pathogens.62–64 There is considerable regional variability with a number of studies showing no association with Chlamydia.65–67 There may be different pathogens in different geographic regions predisposing to the development of ocular adnexal MALT lymphoma. Chlamydia causes chronic infections with inhibition of apoptosis and tumorigenic immunomodulatory effects that predispose to lymphoma formation.68 The chronic systemic infection may be present for years, providing long-term chronic antigen stimulation. The pathogen may elaborate antigens that lead to molecular mimicry, allowing the organism to be tolerated, and other factors contribute to chronic antigen stimulation of both humoral and cell-mediated responses, creating an environment suitable for development of ocular adnexal EMZL. Analysis of the mutations in VH gene segments also suggests chronic antigen stimulation plays a role in ocular adnexal EMZL development.38,69 Decaudin et al. showed a significant association between gastric H. pylori infection and ocular adnexal EMZL. They postulate that lymphocytes undergoing lymphoma-genic change caused by bacterial inflammation at one extranodal site (e.g., gastric) can home to the ocular adnexal region.70,71
Immunosuppression
Immunosuppression has long had been associated with lymphoma development that was underlined by the increased incidence of lymphoma paralleling the acquired immunodeficiency syndrome (AIDS) era. Lymphoma associated with immunosuppression tends to have a high prevalence of Epstein-Barr virus (EBV), defects in immunoregulation, and abnormal immunoglobulin and T-cell receptor gene rearrangement during lymphopoiesis.72 Disorders of immunity such as primary congenital immune deficiency, ataxia telangiectasia, Wiscott-Aldridge syndrome, and therapeutic immunosuppression all predispose to lymphoma development. Interestingly, environments with high ultraviolet (UV) light exposure, such as Australia and the state of Florida in the United States, have high rates of both nonmelanoma skin cancer and lymphoma, suggesting a common role in immunosuppression from UV light in these tumors.73
Pathology
As we enter the era of targeted therapy, we need to establish lymphoma diagnosis accurately. This is best performed with open biopsy, with tissue taken fresh for flow cytometry and in formalin for paraffin-fixed sections for routine histology, morphology, and immunohistochemistry (Fig. 15.7). Fine-needle biopsy can permit diagnosis of lymphoma but does not allow for morphologic assessment, which is important for subtyping the lymphomas.

OAL of MALT type resembles MALT lymphoma elsewhere, comprising morphologically small marginal zone cells, monocytoid cells, and occasional immunoblasts, centroblasts, and small lymphocytes (Fig. 15.8A). There may be some plasmacytic differentiation and infiltration of epithelial tissues with malignant cells to form so-called lymphoepithelial units. Dutcher bodies (PAS-positive pseudointranuclear inclusions) are seen in about 25% of cases (Fig. 15.8B). Immunohistochemically, the cells are CD20 and CD79a positive and CD5 (95%), CD10, and CD23 negative (Fig. 15.8C).74,75

FL recapitulates the normal follicle formation with tumor cells but with poor definition, absence of mantle zone, and effacement of normal architecture with tumor cells (Fig. 15.9). These cells are of two types: small cleaved centrocytes and larger noncleaved centroblasts. They are positive for pan B- cell markers CD 19, CD 20, CD 22, and CD 79a, are Bcl2 positive and express germinal center markers BCL 6, CD38, and CD10, but are CD5 and CD43 negative.47,76

DLBCL diffusely involves tissues with a monotonous proliferation of large neoplastic B cells with large nuclei, which most commonly resemble centroblasts (Fig. 15.10). Again, they are usually positive for pan–B-cell markers CD19, CD20, and CD79a and may be CD5 positive and express germinal center markers CD10 and Bcl-6 and activation markers MUM1 and CD138 variably. They do not express cyclin D1, in contrast to MCL. Importantly, immunohistochemistry can distinguish between the good prognosis CD10 and Bcl-6-positive lesions (GCB DLBCL) and the poorer prognosis activated non-GC DLBCL tumors, which express activation markers MUM1 and CD138 but not CD10 or Bcl-6.77–80

MCL can show somewhat nodular patterns but with loss of normal architecture and infiltration by abnormal centrocyte-like cells, without blast forms (Fig. 15.11). They have a characteristic immunophenotype with CD5 positive and CD43 positive, and BCL6 and CD10 negative. They are all bcl-2 and cyclin D1 positive.51,52

T-cell lesions include a broad range of subtypes but are typically CD20 negative and CD 3 positive, CD30 positive, and ALK variable.81
Features of these subtypes of lymphoma are summarized in Table 15.1.
Table 15.1
Clinical, Pathologic, and Genetic Features of Ocular Adnexal B-Cell Lymphomas
| Type of Lymphoma | Clinical Growth | Clinical Features | Presumed Cell Source | Cell Morphology | Immunoperoxidase Stains | Genetic Abnormalities |
| Hodgkin lymphoma | Contiguous spread | Prognosis based on stage | Germinal center B cells | Reed-Sternberg cells | CD 15, CD 30 + | |
| Non-Hodgkin lymphoma | Prognosis based on grade | (See list below.) | ||||
| Extranodal zone marginal (MALToma) | Indolent | Associated with inflammation (infections/immune) Possible transformation to DLBC | Postfollicular small marginal zone B cells | Heterogeneous infiltrate, including plasmacytes, small lymphocytes, and centrocyte cells | CD 20, CD 5+ | t(11;18) in 10% |
| Follicular lymphoma | Indolent | Possible transformation to DLBC | Germinal center B cells | Back-to-back follicles, monotonous cells (small cleaved centrocytes, large-noncleaved centroblasts) | BCL2+ BCL 6+ CD 20 CD 19+ | t(14;18) + |
| Mantle cell lymphoma | Aggressive | Often poor response to therapy Does not transform to DLBC | Mantle zone B cells | Diffuse infiltrate of small monotonous mantle cells, small cleaved centrocytes | Cyclin D1+ CD 5+ | t(11;14) |
| Diffuse large B-cell (DLBC) lymphoma | Aggressive | May be associated with AIDS, EBV; 35% from follicular lymphoma transformation | Centroblastic vs immunoblastic | Large nuclei over twice normal size | BCL6+ | t(3;14) |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



{kind=link}
{kind=link}