4
Iris Anomalies
Michael J. Bartiss and Bruce M. Schall 
CENTRAL PUPILLARY CYSTS (PUPILLARY MARGIN EPITHELIAL CYSTS)
Etiology
 Usually congenital in origin
Usually congenital in origin
 Can be acquired from cholinesterase inhibiting eye drops, such as phospholine iodide, when used in young, phakic patients to treat accommodative esotropia
Can be acquired from cholinesterase inhibiting eye drops, such as phospholine iodide, when used in young, phakic patients to treat accommodative esotropia
 Rarely inherited
Rarely inherited
Symptoms
 Patients are usually asymptomatic.
Patients are usually asymptomatic.
 Pigmented epithelial cysts occurring at the pupillary border (Fig. 4-1)
Pigmented epithelial cysts occurring at the pupillary border (Fig. 4-1)
 May be detected by pediatrician on red reflex testing of neonate
May be detected by pediatrician on red reflex testing of neonate
Signs
 Pigmented cysts along the margin of the pupil of involved eyes
Pigmented cysts along the margin of the pupil of involved eyes
 Have a nontransparent lining (as opposed to iris stromal cysts)
Have a nontransparent lining (as opposed to iris stromal cysts)
 Rarely increase in size and typically remain stationary
Rarely increase in size and typically remain stationary
Differential Diagnosis
 Iris stromal cysts
Iris stromal cysts
 Ciliary body cysts
Ciliary body cysts
 Iris melanoma
Iris melanoma
Treatment
 Congenital pupillary margin epithelial cysts rarely require treatment; they usually remain stationary in size or slowly involute over time.
Congenital pupillary margin epithelial cysts rarely require treatment; they usually remain stationary in size or slowly involute over time.
 If size and location cause visual compromise, surgical intervention may be indicated.
If size and location cause visual compromise, surgical intervention may be indicated.
 Acquired pupillary margin cysts from cholinesterase-inhibiting eye drops can be prevented with the use of daily phenylephrine (2.5%) eyedrops.
Acquired pupillary margin cysts from cholinesterase-inhibiting eye drops can be prevented with the use of daily phenylephrine (2.5%) eyedrops.
Prognosis
 Excellent; rarely require treatment
Excellent; rarely require treatment
 Complications can include formation of iris flocculi in cases of cyst rupture, glaucoma, and spontaneous intraocular detachment of the cysts.
Complications can include formation of iris flocculi in cases of cyst rupture, glaucoma, and spontaneous intraocular detachment of the cysts.
 If treatment is required, can be treated with simple excision or yttrium aluminium garnet (YAG) puncture
If treatment is required, can be treated with simple excision or yttrium aluminium garnet (YAG) puncture
REFERENCES
Shields JA, Kline MW, Augsburger JJ. Primary iris cysts: a review of the literature and report of 62 cases. Br J Ophthalmol. 1984;68(3):152–166.
Shields JA, Shields CL, Lois N, et al. Iris cysts in children: classification, incidence and management: the 1998 Torrence A Makley, Jr. lecture. Br J Ophthalmol. 1999;83(3):334–338.
Sidoti PA, Valencia M, Chen M. Echographic evaluation of primary cysts of the iris pigment epithelium. Am J Ophthalmol. 1995;120:161–167.
FIGURE 4-1. Pigmented cysts along the margin of the pupil. (Courtesy of Judith Lavrich, MD.)
ANIRIDIA
Etiology
 Bilateral disorder characterized by underdevelopment (rather than true absence of the iris) with rudimentary iris located peripherally
Bilateral disorder characterized by underdevelopment (rather than true absence of the iris) with rudimentary iris located peripherally
 Associated with PAX6 gene (control gene for eye morphogenesis) on chromosome 11p13: involving inability of single gene allele to activate transduction of developmental genes (haploinsufficiency)
Associated with PAX6 gene (control gene for eye morphogenesis) on chromosome 11p13: involving inability of single gene allele to activate transduction of developmental genes (haploinsufficiency)
 Often associated with foveal hypoplasia, nystagmus, glaucoma, optic nerve hypoplasia, cataracts, and acquired corneal pannus
Often associated with foveal hypoplasia, nystagmus, glaucoma, optic nerve hypoplasia, cataracts, and acquired corneal pannus
 Autosomal dominant (complete penetrance with variable expressivity), autosomal recessive (Gillespie’s syndrome with mental retardation and cerebellar ataxia), and sporadic inheritance patterns
Autosomal dominant (complete penetrance with variable expressivity), autosomal recessive (Gillespie’s syndrome with mental retardation and cerebellar ataxia), and sporadic inheritance patterns
 Two-thirds of children with aniridia have affected parents
Two-thirds of children with aniridia have affected parents
 Sporadic aniridia is associated with an increased incidence of Wilms’ tumor.
Sporadic aniridia is associated with an increased incidence of Wilms’ tumor.
 WAGR complex (Wilms’ tumor, aniridia, genitourinary malformations, and mental retardation) occurs from contiguous gene deletions.
WAGR complex (Wilms’ tumor, aniridia, genitourinary malformations, and mental retardation) occurs from contiguous gene deletions.
Symptoms
 Clinical absence of the iris
Clinical absence of the iris
 Subnormal visual acuity common (usually less than 20/100)
Subnormal visual acuity common (usually less than 20/100)
 Nystagmus
Nystagmus
 Photophobia
Photophobia
Signs
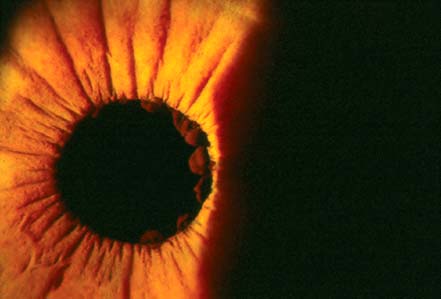
 Apparent bilateral absence or severe hypoplasia of iris (Fig. 4-2)
Apparent bilateral absence or severe hypoplasia of iris (Fig. 4-2)
 Congenital nystagmus
Congenital nystagmus
 Acquired corneal pannus
Acquired corneal pannus
 Strabismus
Strabismus
 Cataract
Cataract
 Ectopia lentis
Ectopia lentis
 Glaucoma
Glaucoma
 Posterior synechiae
Posterior synechiae
Differential Diagnosis
 Other causes of pupillary dilation (e.g., pharmacologically dilated pupils, Aides pupil)
Other causes of pupillary dilation (e.g., pharmacologically dilated pupils, Aides pupil)
Treatment
 Evaluation with a geneticist
Evaluation with a geneticist
 Screening for Wilms’ tumor includes abdominal ultrasonography evaluations every 3 months until age 7 to 8 years of age
Screening for Wilms’ tumor includes abdominal ultrasonography evaluations every 3 months until age 7 to 8 years of age
 Screen for glaucoma and treat if present.
Screen for glaucoma and treat if present.
 Cataract surgery if visually significant cataract is present
Cataract surgery if visually significant cataract is present
 Maximize visual potential with appropriate refractive error correction.
Maximize visual potential with appropriate refractive error correction.
 Polarized sun wear or use of Transitions spectacles lenses to decrease glare and photophobia
Polarized sun wear or use of Transitions spectacles lenses to decrease glare and photophobia
REFERENCES
Adeoti CO, Afolabi AA, Ashaye AO, et al. Bilateral sporadic aniridia: review of management. Clin Ophthalmol. 2010;4:1085–1089.
Lee H, Meyers K, Lanigan B, et al. Complications and visual prognosis in children with aniridia. J Pediatr Ophthalmol Strabismus. 2010;47(4):205–210.
FIGURE 4-2. Severe hypoplasia of iris with outline of lens visible. (Courtesy of Alex V. Levin, MD, MHSc, Wills Eye Institute, Philadelphia.)
BRUSHFIELD SPOTS
Etiology
 Occur in up to 90% of patients with Down’s syndrome (trisomy 21)
Occur in up to 90% of patients with Down’s syndrome (trisomy 21)
 Can be present in patients without Down’s syndrome
Can be present in patients without Down’s syndrome
Symptoms
 Patients are asymptomatic.
Patients are asymptomatic.
Signs
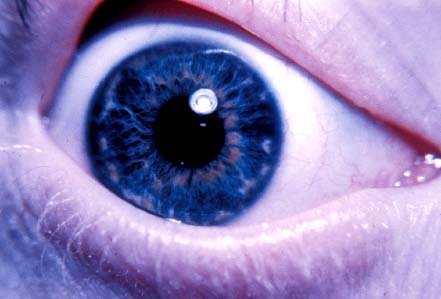
 Whitish elevated spots on the anterior surface of the iris, often occurring in a concentric ring around the pupil (Fig. 4-3)
Whitish elevated spots on the anterior surface of the iris, often occurring in a concentric ring around the pupil (Fig. 4-3)
 Congenital, normal, to hypercellular hypopigmented areas of iris tissue with surrounding relative stromal hypoplasia
Congenital, normal, to hypercellular hypopigmented areas of iris tissue with surrounding relative stromal hypoplasia
Differential Diagnosis
 Wolfflin nodules (similar appearing nodules occurring in patients without Down’s syndrome, which are accumulations of fibrous tissue in the anterior border layer of the iris)
Wolfflin nodules (similar appearing nodules occurring in patients without Down’s syndrome, which are accumulations of fibrous tissue in the anterior border layer of the iris)
 Iris nevi
Iris nevi
 Brushfield’s spots
Brushfield’s spots
 Juvenile xanthogranuloma (JXG)
Juvenile xanthogranuloma (JXG)
 Iris mamillations
Iris mamillations
Treatment
 No treatment indicated
No treatment indicated
Prognosis
 No effect on visual function
No effect on visual function
 Severity of functional cognitive impairment of patients with trisomy 21 is extremely variable.
Severity of functional cognitive impairment of patients with trisomy 21 is extremely variable.
REFERENCES
Brooke Williams RD. Brushfield spots and Wolfflin nodules in the iris: an appraisal in handicapped children. Dev Med Child Neurol. 1981;23(5):646–649.
Shapiro BL. Down syndrome and associated congenital malformations [review]. J Neural Transm Suppl. 2003;(67):207–214.
FIGURE 4-3. Hypopigmented elevated spots on the anterior iris surface in a concentric ring around the pupil. (Courtesy of Alex V. Levin, MD, MHSc, Wills Eye Institute, Philadelphia.)
ECTOPIA LENTIS ET PUPILLAE
Etiology
 Autosomal recessive inherited, nonprogressive disorder in which the pupil and lens are displaced in opposite directions (pupil usually inferonasally and lens superotemporally)
Autosomal recessive inherited, nonprogressive disorder in which the pupil and lens are displaced in opposite directions (pupil usually inferonasally and lens superotemporally)
 Posterior displacement of lens–iris diaphragm
Posterior displacement of lens–iris diaphragm
 Typically bilateral and asymmetric
Typically bilateral and asymmetric
 Believed to occur during neuroectodermal tissue development (pigmented layers of iris, iris dilator, and zonules are all involved)
Believed to occur during neuroectodermal tissue development (pigmented layers of iris, iris dilator, and zonules are all involved)
Symptoms
 Decreased uncorrected visual acuity secondary dislocated lens
Decreased uncorrected visual acuity secondary dislocated lens
Signs
 Bilateral lens dislocation causing high myopia with astigmatism
Bilateral lens dislocation causing high myopia with astigmatism
 Asymmetrical, eccentrically located pupils (usually inferonasally)
Asymmetrical, eccentrically located pupils (usually inferonasally)
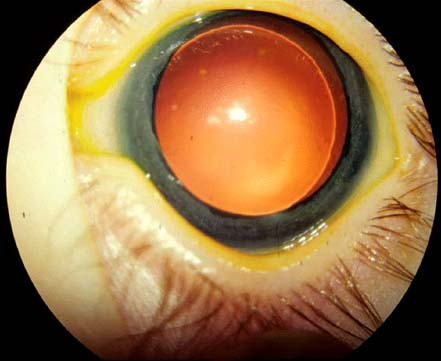
 Slit-shaped or oval pupil (Fig. 4-4A)
Slit-shaped or oval pupil (Fig. 4-4A)
 Persistent pupillary membrane is present in approximately 85% of affected individuals (Fig. 4-4B)
Persistent pupillary membrane is present in approximately 85% of affected individuals (Fig. 4-4B)
 Microspherophakia, miosis, and poor dilation with mydriatic agents
Microspherophakia, miosis, and poor dilation with mydriatic agents
 Myopia, which may be severe
Myopia, which may be severe
 May have an enlarged corneal diameter
May have an enlarged corneal diameter
 Cataract
Cataract
 Abnormal iris transillumination
Abnormal iris transillumination
 Retinal detachment
Retinal detachment
 +/– Megalocornea
+/– Megalocornea
Differential Diagnosis
 Other causes for bilateral dislocated lenses, Marfan’s syndrome, homocystinuria, Weil-Marchesani syndrome, sulfite oxidase deficiency, hyperlysinemia
Other causes for bilateral dislocated lenses, Marfan’s syndrome, homocystinuria, Weil-Marchesani syndrome, sulfite oxidase deficiency, hyperlysinemia
 Iris coloboma
Iris coloboma
 Trauma to iris sphincter
Trauma to iris sphincter
 After anterior segment surgery
After anterior segment surgery
 Corectopia
Corectopia
 Axenfeld-Rieger syndrome
Axenfeld-Rieger syndrome
Treatment
 Refractive error correction to maximize visual potential
Refractive error correction to maximize visual potential
 Anisometropic amblyopia often occurs in the more affected eye. Amblyopia should be treated with correction of refractive and occlusion of fellow eye.
Anisometropic amblyopia often occurs in the more affected eye. Amblyopia should be treated with correction of refractive and occlusion of fellow eye.
 May develop a visually significant cataract and therefore may require cataract surgery
May develop a visually significant cataract and therefore may require cataract surgery
 Screen for glaucoma.
Screen for glaucoma.
Prognosis
 Nonprogressive; visual prognosis depends on timely treatment of refractive error. Amblyopia in more affected eye is often severe and may not respond to treatment.
Nonprogressive; visual prognosis depends on timely treatment of refractive error. Amblyopia in more affected eye is often severe and may not respond to treatment.
REFERENCES
Byles DB, Nischal KK, Cheng H. Ectopia lentis et pupillae. A hypothesis revisited. Ophthalmology. 1998;105(7):1331–1336.
Colley A, Lloyd IC, Ridgway A, et al. Ectopia lentis et pupillae: the genetic aspects and differential diagnosis. J Med Genet. 1991;28(11):791–794.
Goldberg MF. Clinical manifestations of ectopia lentis et pupillae in 16 patients. Ophthalmology. 1988; 95(8):1080–1087.
FIGURE 4-4. A. Inferonasally eccentrically located slit-shaped pupil. (Courtesy of Alex V. Levin, MD, MHSc, Wills Eye Institute, Philadelphia.) B. Note the persistent pupillary membrane, which is being stretched in this pharmacologically dilated pupil. The superior edge of the dislocated lens is visible.
HETEROCHROMIA IRIDIS
Etiology
 A congenital or acquired condition characterized by a relative hyperpigmentation or hypopigmentation of the involved iris
A congenital or acquired condition characterized by a relative hyperpigmentation or hypopigmentation of the involved iris
 Acquired cases of hyperpigmented irides in children include trauma, siderosis, iris ectropion syndrome, chronic iridocyclitis, and extensive rubeosis as well as intraocular surgery and topical prostaglandin analogue medications
Acquired cases of hyperpigmented irides in children include trauma, siderosis, iris ectropion syndrome, chronic iridocyclitis, and extensive rubeosis as well as intraocular surgery and topical prostaglandin analogue medications
 Ocular melanocytosis or oculodermal melanocytosis and sector iris hamartoma can also cause hyperpigmented irides.
Ocular melanocytosis or oculodermal melanocytosis and sector iris hamartoma can also cause hyperpigmented irides.
 Congenital and acquired hypopigmented irides can occur because of Horner’s syndrome, Fuchs heterochromia, Waardenburg-Klein syndrome, nonpigmented iris tumors, and hypomelanosis of Ito.
Congenital and acquired hypopigmented irides can occur because of Horner’s syndrome, Fuchs heterochromia, Waardenburg-Klein syndrome, nonpigmented iris tumors, and hypomelanosis of Ito.
Symptoms
 Patients are typically asymptomatic in the absence of rubeosis, increased intraocular pressure (IOP), and intraocular inflammation.
Patients are typically asymptomatic in the absence of rubeosis, increased intraocular pressure (IOP), and intraocular inflammation.
Signs
 Different-colored irides with or without anatomical iris abnormalities
Different-colored irides with or without anatomical iris abnormalities
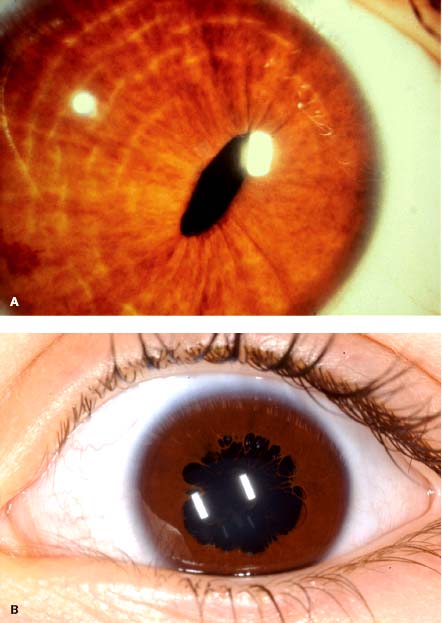
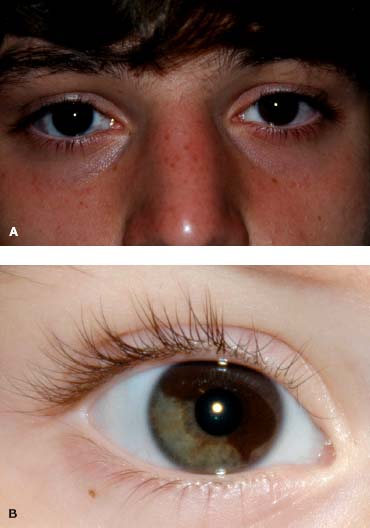
 In cases of melanosis oculi, the more pigmented iris may appear thicker with mamillations (Fig. 4-5).
In cases of melanosis oculi, the more pigmented iris may appear thicker with mamillations (Fig. 4-5).
 Associated with miosis and ptosis (typically 2 mm) on the ipsilateral side in cases of Horner’s syndrome (Fig. 4-6)
Associated with miosis and ptosis (typically 2 mm) on the ipsilateral side in cases of Horner’s syndrome (Fig. 4-6)
Differential Diagnosis
 Differential diagnosis is extensive.
Differential diagnosis is extensive.
 Acquired cases of hyperpigmented irides in children include trauma, siderosis, iris ectropion syndrome, chronic iridocyclitis, and extensive rubeosis as well as intraocular surgery and topical prostaglandin analog medications.
Acquired cases of hyperpigmented irides in children include trauma, siderosis, iris ectropion syndrome, chronic iridocyclitis, and extensive rubeosis as well as intraocular surgery and topical prostaglandin analog medications.
 Ocular melanocytosis or oculodermal melanocytosis and sector iris hamartoma can also cause hyperpigmented irides.
Ocular melanocytosis or oculodermal melanocytosis and sector iris hamartoma can also cause hyperpigmented irides.
 Congenital and acquired hypopigmented irides can occur because of Horner’s syndrome, Fuchs heterochromia, Waardenburg-Klein syndrome, nonpigmented iris tumors, and hypomelanosis of Ito.
Congenital and acquired hypopigmented irides can occur because of Horner’s syndrome, Fuchs heterochromia, Waardenburg-Klein syndrome, nonpigmented iris tumors, and hypomelanosis of Ito.
 Neuroblastoma (located along the sympathetic chain) must be ruled out in cases of Horner’s syndrome, especially in acquired cases in children.
Neuroblastoma (located along the sympathetic chain) must be ruled out in cases of Horner’s syndrome, especially in acquired cases in children.
Treatment
 Assessment as to which iris has the abnormal color can often be assisted by assessing skin pigmentation, parental eye color, and earlier photographs of the patient.
Assessment as to which iris has the abnormal color can often be assisted by assessing skin pigmentation, parental eye color, and earlier photographs of the patient.
 Timely and appropriate workup of acquired Horner’s syndrome
Timely and appropriate workup of acquired Horner’s syndrome
 Hearing testing if Waardenburg’s syndrome is suspected
Hearing testing if Waardenburg’s syndrome is suspected
 In cases of acquired hyperpigmentation, imaging may be needed to rule out an intraocular foreign body (siderosis) and intraocular tumor.
In cases of acquired hyperpigmentation, imaging may be needed to rule out an intraocular foreign body (siderosis) and intraocular tumor.
Prognosis
 Depends on the underlying cause
Depends on the underlying cause
REFERENCES
Brazel SM, Sullivan TJ, Thorner PS, et al. Iris sector heterochromia as a marker for neural crest disease. Arch Ophthalmol. 1992;110(2):233–235.
Liu XZ, Newton VE, Read AP. Waardenburg syndrome type II: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995 2;55(1):95–100.
Milunsky JM. Waardenburg syndrome type I. In: Pagon RA, Bird TC, Dolan CR, Stephens K, eds. GeneReviews. Seattle: University of Washington; 2004.
FIGURE 4-5. A. Right iris is more heavily pigmented in this child with melanosis oculi. B. Sector melanosis oculi.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


