5
Inner Ear Molecular Disorders
Joni K. Doherty, David J. Lim, and Rick A. Friedman
Much of what we know about the workings of the inner ear at the molecular level has been attained through analysis of mutations that result in hearing and balance dysfunction. Dissection of a particular mutation to determine its genomic location and gene product, including structure, subcellular location, and function, can provide vast information in terms of inner ear molecular mechanisms. Genetic mutations affecting the inner ear are being intensely studied in both humans and animal models, resulting in a dramatic increase in our understanding of the molecular biology of the inner ear. This chapter reviews the current state of our understanding based on such studies.
 Identification of Deafness Genes in Human Studies
Identification of Deafness Genes in Human Studies
Hearing loss (HL) is the most common sensory impairment in humans. At least one out of every 750 babies born in the United States has sensorineural HL (SNHL). Demographic studies estimate that approximately 50% of all congenital deafness can be traced to inheritable factors—most due to single gene mutations—and at least 90% are inherited as autosomal recessive (AR) traits. Of these, approximately 30% are syndromic and 70% are nonsyndromic. Considerable progress has been made in identifying and characterizing these deafness genes.
During the 1980s, advances in molecular genetic technology led to a rapidly progressive increase in the number of molecular markers that facilitate linkage analysis. These markers, or polymorphisms, define genetic locations and have enabled scientists to identify the approximate location of inherited mutations. Thus, gene mapping began to be applied to familial deafness in the late 1980s, resulting in the first definitive information regarding the physical site and nature of the mutations affecting the inner ear. The recent development of thousands of molecular tags for specific locations in the genome has dramatically enhanced the ability to link inherited disorders to their specific chromosomal location via recombination frequency analysis. When combined with information from prior localization studies of candidate genes or related animal mutations in the homologous chromosomal region, such linkage analysis has enabled the characterization of an increasing number of mutations that cause disorders of hearing. Approximately 200 deafness loci have been identified, including syndromic and nonsyndromic forms. Our knowledge of the identity and function of these genes has grown almost exponentially over the last decade. Today, more than 10 genetic mutations leading to congenital hearing loss can be identified with genetic testing (go to www.genetests.com for testing laboratory information). This has improved our ability to provide genetic counseling, and, in the future, may lead to gene therapy for otologic diseases. Gene identification also allows for determination of function via animal studies of homologous gene mutations that can be genetically engineered.
Syndromic Deafness
Genetic mutations causing congenital deafness in association with other morphologic or clinical features are referred to as syndromic deafness genes. Syndromic deafness accounts for 30% of inherited congenital hearing loss.1 Linkage analysis has been facilitated by the fact that affected individuals are more easily distinguishable from unaffected members within a given population or family with respect to a specific syndrome. The specific mutations causing Alport syndrome, Waardenburg syndrome, Usher syndrome, branchio-oto-renal (BOR) syndrome, Pendred syndrome, X-linked deafness with perilymph gusher, neurofibromatosis type 2 (NF2), and many other forms of syndromic deafness, as well as nonsyndromic deafness, have been determined (Fig. 5–1), and many have been characterized, in terms of the molecular defect (Table 5–1). The mitochondrial DNA mutation leading to inherited susceptibility to aminoglycoside-induced hearing loss has also been identified.2 Here, we will discuss the molecular genetics of inherited deafness. Chapter 22 discusses the clinical aspects of these syndromes in more detail.
Alport Syndrome
The first human mutation affecting the inner ear to be identified was that causing Alport syndrome, an X-linked disease associated with renal abnormalities and progressive HL. The mutated gene was initially linked to Xq22–26 and was subsequently identified as that encoding the α5 chain of type IV collagen, which plays a critical role in the formation of basement membranes.3 Six subtypes of Alport syndrome have since been identified: types II, III, and IV are X-linked, whereas types I, V, and VI are inherited in an autosomal dominant (AD) fashion.1
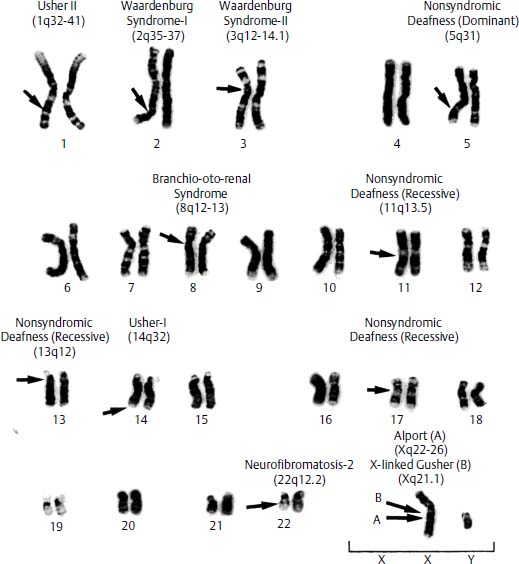
Figure 5–1 Human male karyotype with chromosomal locations of a few of the known deafness genes, including syndromic and one nonsyndromic loci. Arrows indicate the approximate location of each of the genes on each particular chromosome.
Waardenburg Syndrome
Waardenburg syndrome (WS) can be inherited as an AD, AR, or sporadic mutation. WS type I (WS-I) is characterized by hearing loss and pigmentary abnormalities such as white forelock and heterochromia irides. Waardenburg syndrome type II (WS-II) is distinguished from WS-I by the presence of dystopia canthorum, which presents as lateral displacement of the medial canthi. The AD WS-I defect was initially localized to the long arm of chromosome 2 (2q35–37) because of a de novo chromosomal malformation,4 and then further linked to a gene near 2q37.5 This locus is homologous to the Pax3 gene in Splotch mice, which had been proposed as a murine equivalent of WS-I (Fig. 5–2). Mutations in the PAX3 gene of WS-I patients were subsequently shown to be responsible for WS-I and WS type III (WS-III).6 PAX3 encodes a transcription factor (TF) that regulates expression of other genes. The PAX3 protein product plays a regulatory role in melanocyte differentiation during embryogenesis when they migrate from neural crest to various tissues, including the intermediate cells of the stria vascularis in the cochlea and the melanocytes apposing the dark cells of the vestibular labyrinth, where they are necessary for production of endolymph and the endolymphatic potential. In VGA-9 mice, harboring a transgenic mutation of Pax3 similar to that in Splotch mice (the murine equivalent of WS-I), the stria vascularis shows a lack of intermediate cells and disorganization of the basal layer (Fig. 5–2). WS-II has not shown linkage to the region of chromosome 2 that includes PAX3, and WS-II appears to be caused by mutation of either the microphthalmia-associated TF, MITF, which maps to 3p12.3–14.1, or SNA12, which is located at 8q11 and encodes the SLUG transcription factor.1 Type IV Waardenburg (WS-IV) maps to still other sites: (1) 20q13.2–3, where the EDN3 gene is located, encoding endothelin-37; (2) 13q22, where ENDRB encodes the endothelin receptor, type B; and (3) 22q13, encoding the transcription factor SOX10. Endothelin signaling is involved in neuronal survival, and patients with WS-IV develop Hirschsprung disease (lack of autonomic innervation to the distal large colon) in addition to signs and symptoms of WS-II.8
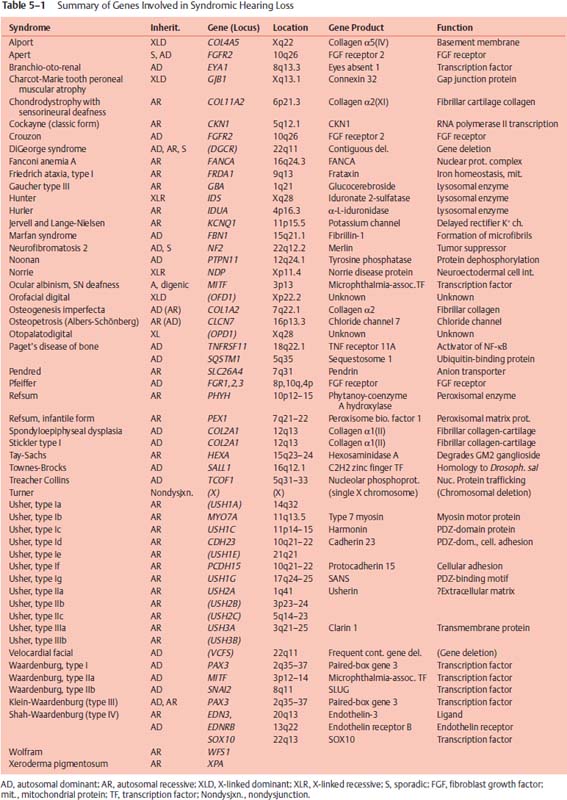
Figure 5–2 Loss of intermediate cells in the stria vascularis of transgenic VGA-9 mice, a murine equivalent of Waardenburg syndrome type I (WS-I) and very similar to Splotch mice. (A) There are three cell layers in the normal (NL) stria: basal (b), intermediate (i), and marginal (m). (B) Note the absence of the intermediate cells and the disorganization of the basal layer in the homozygous (HO) mouse. The marginal cells do not interdigitate with the basal cells and appear somewhat abnormal with larger, more cuboidal nuclei. Original magnification ×63.
Usher Syndrome
Usher syndrome (US) is inherited in an AR fashion and is characterized by deafness and retinitis pigmentosa. Usher syndrome type I (US-I) is characterized by severe HL and impaired vestibular function, whereas Usher syndrome type II (US-II) has less severe involvement of both. Additional subtypes based on severity and character of progression have been proposed: US-III and US-IV. Both US-I and US-II are genetically heterogeneous, with some US-I families linking to chromosome 14q32 and others linking to chromosome 11, and the Shaker-1 mouse mutation, which maps to a region homologous to human 14q32, was shown to involve a gene encoding Myosin VII.1 Based on this observation, Myosin VIIA was subsequently identified as the gene responsible for Usher syndrome Ib.10 Although incompletely understood, the role of Myosin VIIA in the inner ear appears to involve inner hair cell (IHC) integrity. Myosin VIIA localizes to the cross-links between adjacent stereocilia and at the cuticular plate of IHCs (Fig. 5–3). It has been postulated to play a role in adaptation, which is the process of maintenance of lateral tip link tension and restoration of hair cell sensitivity after depolarization.11–13 Myosin VIIA can move upward on the actin core within stereocilia, and may connect with cadherin-catenin complexes via the PDZ domain-containing protein harmonin in an adaptation motor to maintain stereocilia tension as well as lateral link tension within hair cells (HCs) (Fig. 5–3). Mutations in harmonin cause Usher syndrome Ic.14 US-IIa links to chromosome 1q41, where the gene product, named Usherin, is encoded. Usherin contains laminin-epidermal growth factor (EGF) and fibronectin domains, and thus likely functions in extracellular matrix interactions.1,15–18 Overall, at least nine distinct chromosomal locations have been linked to the various forms of US.1,9
Pendred Syndrome
Pendred syndrome is an AR disorder characterized by deafness and euthyroid goiter that results from mutation of the PDS gene, also called SLC26A4, which encodes the pendrin protein. Pendrin is a putative ion transport molecule that appears to function as an iodide (in the thyroid), chloride, bicarbonate, formate, and nitrate transporter.19 Pendrin is discretely expressed in (1) the apical membrane of endolymphatic sac cells; (2) the transitional cells of the cristae ampullaris, utriculi, and sacculi; (3) on the apical surface of basal and intermediate cells within the stria vascularis in the cochlea of the embryonic mouse; and (4) in the apical membrane of cells in the spiral prominence and along the root processes of outer sulcus cells in the cochlea.20 Based on studies of PDS knockout mice, pendrin appears to play a vital role in endolymph fluid homeostasis in the inner ear as well as maintenance of the endolymphatic potential.20 The spectrum of inner ear abnormalities associated with PDS mutations ranges from isolated enlarged vestibular aqueduct (EVA) to classic Mondini malformation, or incomplete partition type II, with cystic apical and middle turns of the cochlea, lacking the interscalar septum, a mildly dilated vestibule, and EVA. Hearing is similarly variable, ranging from normal in early childhood to mild low-frequency conductive loss to profound SNHL. Stepwise progression of hearing loss is the typical pattern, and there have been no reported cases with normal hearing beyond early childhood. In association with Pendred syndrome, euthyroid goiter and a positive perchlorate discharge test are present.
Neurofibromatosis Type 2
Affecting 1 in 40,000 individuals, neurofibromatosis type 2 (NF2) is caused by a mutation at 22q12.2.21 Individuals affected with NF2 characteristically develop bilateral vestibular nerve schwannomas, inevitably leading to HL and balance dysfunction. NF2 mutation is also associated with schwannomas of other cranial nerves, meningiomas, and spinal tumors. NF2 is inherited in an AD fashion, but approximately 50% of cases are sporadic. Mosaicism of NF2 gene mutation also occurs in up to 25% of cases. The NF2 gene encodes merlin, or schwannomin, an intracellular tumor suppressor protein expressed in Schwann cells. Merlin shares extensive homology with the erzin/radixin/moesin family, is negatively regulated by phosphorylation-mediated by p21-activated kinase 1 (PAK1) or focal adhesion kinase (FAK), and translocates to the cytoplasmic cell membrane in a paxillin-dependent manner, where it binds to ErbB2.22 ErbB2 is a growth factor receptor tyrosine kinase that couples with ErbB3 when stimulated by neuregulin binding and activates intracellular signaling cascades, such as mitogen-activated protein kinase (MAPK) and pAkt/PI3-K, thus mediating Schwann cell proliferation and survival. Merlin also has been shown to translocate to the nucleus where it may affect gene expression in Schwann cells.22 However, the role of merlin as a tumor suppressor gene is still poorly understood.
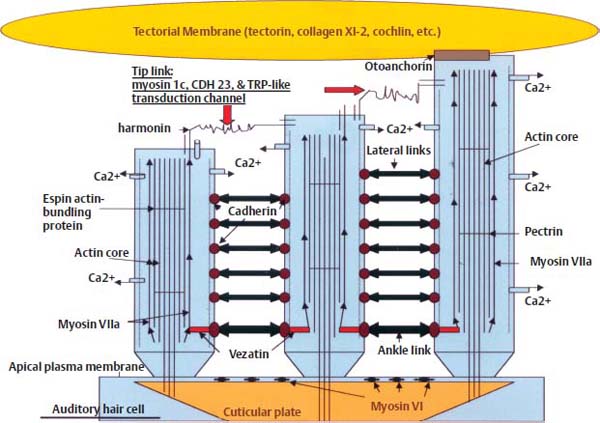
Figure 5–3 Auditory hair cell schematic diagram illustrating molecules involved in mechanosensory transduction of sound. The stereocilia are anchored into the cuticular plate at the apical aspect of the hair cell. The apical plasma membrane is anchored to the cuticular plate by myosin VI, acting in opposition to myosin VIIa, which is involved in elongation of stereocilia via association with the actin core. The tallest stereocilium is attached to the tectorial membrane via otoanchorin, and is connected by cross-links to the shorter stereocilia. The stereocilia contain a circumferential actin core linked by longitudinal pectrin filaments, and they are tapered at the base, which facilitates pivoting rather than bending motion. Movement results in opening of the transduction channels located at either end of the tip links (composed of myosin 1c, cadherin 23, and TRP-like channels) and linked to the myosin Vlla–actin core via harmonin, which is involved in maintenance of lateral link tension. Vezatin facilitates ankle link tension. (Adapted from Hone SW, Smith RJ. Understanding inner ear physiology at the molecular level. Adv Otorhinolaryngol 2002;61:1–10. Reprinted with permission.)
Other Deafness Syndromes
Apert syndrome is AD or, more commonly, sporadic, and results from mutation of the fibroblast growth factor receptor 2 (FGFR2) gene at 10q26. Congenital stapedial footplate fixation results in conductive HL and other craniofacial deformities as well as developmental delay, which are secondary to craniosynostosis in Apert syndrome.1
Branchio-oto-renal syndrome is AD in inheritance and is characterized by several branchial arch deformities, deafness, and renal anomalies. The mutation has been localized to a gene at 8q13.3, termed EYA1 for the “eyes absent” phenotype associated with the orthologous mutation in Drosophila.23,24
X-linked deafness with a perilymph gusher is an X-linked progressive mixed deafness associated with a perilymph (cerebrospinal fluid) “gusher” during stapes surgery, as the name implies. It was linked to Xq21.1 by examination of cytogenetically visible deletion.25–27 The gene mutation causing X-linked mixed deafness with stapes fixation, or DFN3, has recently been identified as POU3F4, which encodes the TF Brn-4, a member of the POU-domain family that is extensively expressed during cochlear development.28 Although the phenotype associated with this disorder only involves the inner ear abnormality, it has historically been classified as a syndromic form of HL, because there is occasionally a radiographic finding of a bulbous distal internal auditory canal associated with the characteristic stapes gusher.
Other AD syndromic forms of HL include Crouzon, DiGeorge, Goldenhar (oculoauricular vertebral dysplasia), osteogenesis imperfecta, Paget, Noonan, Pfeiffer, Stickler, Townes-Brocks, Treacher Collins, and velocardial facial syndromes. Other AR syndromic HL syndromes include Cockayne, Fanconi anemia A, Friedrich ataxia, Hurler, Jervell and Lange-Nielsen, osteopetrosis (Albers-Schönberg), Refsum, Tay-Sachs, and xeroderma pigmentosum. X-linked syndromic deafness disorders include Charcot-Marie Tooth, Hunter, Norrie, otofacial digital, and otopalatodigital syndromes. Table 5–2 lists the genetic defects associated with these disorders, and Chapter 22 discusses their clinical features in detail. Mitochondrial deafness syndromes are discussed in a separate section of this chapter. For a more complete and current review of syndromic deafness genes, go to the Online Mendelian Inheritance in Man Web site at http://www.ncbi.nlm.nih.gov/Omim/.
Nonsyndromic Deafness
Although 30% of prelingual deafness is syndromic, 70% is nonsyndromic HL (NHL).29 NHL is a diagnosis that applies to patients who suffer genetic HL without associated phenotypic abnormalities, and it results from mutations in an estimated 100 genes. Linkage analysis is very difficult in this group of patients due to the genetic heterogeneity. However, using relatively isolated populations and large families, linkage analysis has facilitated the identification of over 70 loci and cloning of more than 21 genes responsible for NHL. Identification of genetic mutations associated with deafness enables genetic testing, precise diagnosis, and genetic counseling. Human temporal bone studies have allowed observations of morphologic defects resulting from such mutations, providing speculative information of gene function. Animal studies utilizing homologous targeted mutations facilitate experimental studies of gene function, and have been the major tool in expanding our knowledge of inner ear protein functions. Because the number of DNA polymorphic markers for genetic NHL is increasing exponentially, up-to-date information can be found on the Hereditary Hearing Loss Homepage at http://dnalab-www.uia.ac.be/dnalab/hhh.
Molecules Involved in Ion Homeostasis
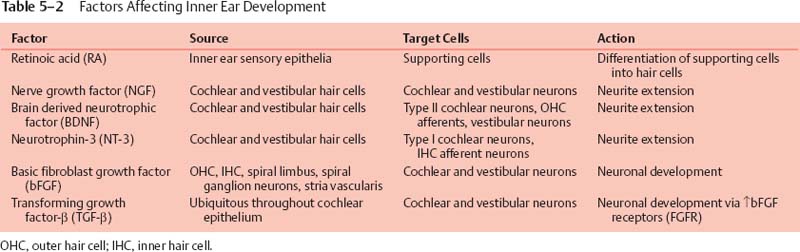
Maintaining the endolymphatic potential within the cochlear duct is essential for normal HC function, and it is postulated that the potassium content in endolymph is highly regulated via a bimodal recycling pathway. According to the predominant theory, potassium is recycled both medially and laterally within the cochlea. The medial pathway involves the spiral limbus interdental cells medial to the organ of Corti, which recycle potassium ions (K+) back into the scala media after auditory HC depolarization (Fig. 5–4); see Chapter 1 for a description of cellular subtypes. The lateral pathway involves the supporting cells adjacent to HCs within the organ of Corti, that recycle K+ through the spiral ligament and stria vascularis back into the scala media (Fig. 5–4). Potassium recycling requires a network of ion channels and transporters, including gap junctions assembled by connexins, the anion transporter pendrin, and potassium channels KCNQ and KCNE.29
Connexins
Currently, GJB2, which encodes connexin 26, is recognized as the most frequent gene mutation associated with congenital deafness in Caucasians. DFNA3, the dominantly inherited form, and DFNB1, the AR mutation, account for up to 50% of cases of congenital deafness. Although relatively small, with the entire coding sequence contained in one exon, more than 50 mutations within the GJB2 gene account for HL. The most common is the 35delG mutation in European countries and the United States.30 In contrast, the 235delC mutation accounts for 73% of GJB2 mutations in the Japanese population.31 The small coding region has facilitated genetic screening methods for GJB2 mutations, which can now be done more rapidly and easily using microarray.32 The role of connexin 26 in the inner ear is related to its function as a subunit of connexon gap junctions. Connexons are composed of hexameric connexin molecules forming a hemipore. The connexon hemipore of two adjacent cells will join to form a complete gap junction and allow intercellular diffusion of K+. Other connexin proteins associated with NHL include connexin 30 (encoded by GJB6, associated with DFNA3 and DFNB1 loci), 31 (DFNA2 locus, GJB3 gene), and 43 (GJA1). Connexins form mostly homomeric connexons, but the heteromeric connexon of connexin 26 and connexin 32 is also expressed in the inner ear. Connexons appear to be essential for generation and maintenance of the endocochlear potential due to their role in K+ recycling (Fig. 5–4).33 There are two independent networks of intercellular connexons that seem to play an important role in recycling of K+ ions: (1) the epithelial network within the basilar membrane adjacent to the organ of Corti both medially and laterally; and (2) the fibrocyte network, which is present within the spiral limbus (medially) and spiral ligament (laterally) and includes the basal, intermediate, and endothelial cells of the stria vascularis. The major pathway for K+ recycling appears to involve the stria vascularis (Fig. 5–4), and connexons have been implicated as part of the structure required for this process, via transcellular circulation of K+ through the spiral ligament and basal cells of the stria vascularis.33
Other Ion Transport Molecules
Another junctional protein important for fluid homeostasis in the inner ear is Claudin 14, a CLDN14 gene product. Claudin 14 forms tight junctions within sensory epithelia of the cochlea and vestibular organs, and NHL results from mutation at the DFNB29 locus (Fig. 5–4).34
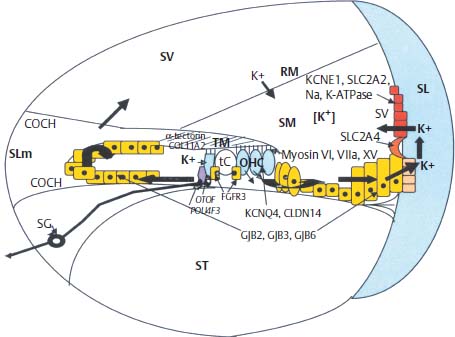
Figure 5–4 Schematic drawing of a cross section through a single turn of the cochlea illustrating potassium recycling pathways medially and laterally (arrows). (See text for description of genes and their role in ion homeostasis; see Chapter 1 for a more detailed description of cochlear anatomy, including cellular subtypes.) SG, spiral ganglia; SLm, spiral limbus; SV, stria vascularis; SL, spiral ligament; SV, scala vestibule; ST, scala tympani; SM, scala media; RM, Reissner’s membrane; K+, potassium ion; TM, tectorial membrane; tC, Corti’s tunnel; OHC, outer hair cell.
PDS (SLC26A4) gene mutations that lead to EVA in isolation (i.e., in absence of goiter or a positive perchlorate discharge test) are a form of nonsyndromic deafness, DFNB4, and are thought to be part of a continuum of diseases caused by the same gene, where Pendred syndrome in association with Mondini malformation (as discussed earlier; see Syndromic Deafness) is at the opposite end of the spectrum.35 Expression studies in Xenopus oocytes as well as mammalian cells, including knockout studies in mice, indicate that pendrin function can support chloride/formate, chloride/hydroxide, chloride/bicarbonate, chloride/nitrate, and chloride/iodide ion exchange.19 Notably, PDS knockout mice develop severe hydrops, suggesting a role for pendrin in endolymphatic fluid homeostasis.
Potassium channels in the inner ear are encoded by the KCNQ1-4 and KCNE1-4 genes. Splice variants from these genes are expressed as subunits of the various isoforms of potassium channels present throughout the mammalian inner ear.36 Together, the KCNE and KCNQ subunits form a functional potassium channel. The KCNE1 (minK) subunit IsK regulates the pore-forming KvLQT1 α-subunit encoded by KCNQ1, which is expressed at the apical surface of marginal cells in the stria vascularis (Fig. 5–4). Mutation in any of these genes can result in deafness, presumably secondary to lack of endocochlear potential generation. These potassium channels are expressed in cardiac tissue as well; Jervell and Lange-Nielsen syndrome results from mutations in either KCNE1 or KCNQ1.36,37 KCNQ4 is expressed by cochlear outer hair cells (OHCs) and vestibular type I HCs. Several lines of evidence suggest that KCNQ4 channels are responsible for the resting K+ current described in OHCs and type I HCs, which likely influence their electrical properties. Furthermore, KCNQ4 is expressed in central auditory pathway nuclei, suggesting that defects in KCNQ4, which lead to DFNA2, contribute to both peripheral and central deafness.38
Transcription Factors
The POU-domain family of transcription factors contains the genes POU4F3 and POU3F4, both of which have been identified in association with late-onset progressive NHL. These TFs are expressed in the late stages of inner ear development, and are important for neuronal differentiation and survival. POU3F4 is expressed in the otic capsule in mesenchyme of the cochlear and the vestibular primordial.39 Mutations in POU3F4 are associated with X-linked progressive mixed HL due to stapes fixation with a progressive SNHL, termed DFN3.25 It is associated with increased perilymphatic (cerebrospinal fluid) pressure and a gusher at stapedectomy, as discussed earlier (see Syndromic Deafness). Conversely, POU4F3 is expressed in developing cochlear and vestibular hair cells.40 A family with a dominantly inherited mutation in the POU4F3 gene encoding a truncated Brn-3.1 resulting in late-onset nonsyndromic sensorineural deafness has been identified, and the deafness gene termed DFNA15.41
Proteins with Unknown Function
A common form of AD low-frequency nonsyndromic SNHL is caused by mutation of the WFS1 gene designated DFNA6/14. WFS1 encodes wolframin, a protein that localizes to the endoplasmic reticulum and is thought to play a role in protein sorting and trafficking, although the function is unknown.42
Cytoskeletal Proteins
Structural components of the cochlea and vestibular organs are highly organized, and their maintenance is essential for the function of hearing and balance, especially with respect to sensory HCs. Mutation in many of the genes that encode cytoskeletal proteins within hair cells leads to NHL. These include (1) a conventional myosin, MYH9; (2) four unconventional myosins, MYO3A, MYO6, MYO7A, and MYO15; (3) stereocilin, a novel stereocilia-associated protein; (4) harmonin, a PDZ-domain protein; (5) a putative actin-polymerization protein, HDIA1; and (6) a cadherin, CDH23.29
Stereocilia are the main structures responsible for mechanosensory transduction in auditory and vestibular HCs. Stereocilia are cellular organelles that are organized into rows of increasing height to create the characteristic staircase pattern seen by electron microscopy. Unlike vestibular HCs, mature auditory hair cells do not contain a kinocilium. Stereocilia are exquisitely sensitive to mechanical vibration, and can easily be damaged by overstimulation, but undergo continuous renewal from tip to base to continue to function an entire lifetime.43 Each stereocilium is composed of a rigid central structure containing several hundred parallel, polarized, and cross-linked actin filaments. Different members of the myosin family are present with these actin filaments in specific locations within stereocilia: myosin XVa (MYO15A gene product) is located at the tips, whereas myosin VIIa (encoded by MYO7A) is located alongside actin at crosslinks of adjacent stereocilia, and myosin VI (gene product of MYO6) is present at the cuticular plate (Fig. 5–3). Myosins bind actin, forming the molecular motor units that generate movement via adenosine triphosphate (ATP) hydrolysis. The MYO7A gene, also the causative gene mutation associated with Usher Ib, is associated with two forms of nonsyndromic deafness: DFNB2, an AR disorder; and DFNA11, an AD disorder.10,44
Harmonin, encoded by USH1C at 11p14–15.2, is associated with NHL via mutations linking to the DFNB18 locus. Harmonin contains a PDZ domain, which suggests that it may function as an assembling protein. PDZ (an acronym designated after three proteins that contain this domain—PSD-95, discs large, and zona occludens—were identified) proteins organize protein complexes into their specific subcellular location, they anchor transmembrane proteins, and recruit cytosolic signaling molecules, and may bind directly to the actin cytoskeleton.14
A putative actin-polymerizing protein is expressed as the gene product of HDIA1, the human homologue of the Drosophila diaphanous gene.45 HDIA1 is a formin gene family member, of which most are involved in cytokinesis and establishing cell polarity.
A novel cadherin-like protein is encoded by CDH23, which is also linked to Usher type 1d, and mutation at the DFNB12 locus results in NHL. Cadherins form adherin junctions, which are critically important during embryogenesis and organogenesis. Stereocilia organization is disrupted early during hair cell differentiation in mouse mutants.46
Synaptic Vesicle Trafficking
OTOF mutation, associated with DFNB9, is reported to be a frequent cause of recessive prelingual NHL in Spanish patients, and accounts for 4.4% of recessive prelingual NHL not due to GJB2 (connexin 26) mutations.47 OTOF encodes otoferlin, which has been identified as a calcium-triggered synaptic vesicle trafficking protein that interacts with syntaxin1 and SNAP25 of the SNARE complex for Ca2+-dependent presynaptic vesicle exocytosis within IHCs. Otoferlin, therefore, appears to be important for afferent neural signaling, and mutations have also been found to be associated with auditory neuropathy.48
Extracellular Matrix Components
Overlying the auditory HCs within the organ of Corti is the tectorial membrane, which generates the shearing force that bends the stereocilia, opening transduction channels and initiating depolarization. This composes the mechanosensory transduction process that is necessary for hearing. Additionally, the tectorial membrane matrix acts as a second resonator and ensures that the OHC bundles are displacement coupled to the sound stimulus. Thus, it facilitates optimal electromechanical feedback to the basilar membrane from OHCs.49 The tectorial membrane consists of collagen fibers and a noncollagenous matrix containing mostly α- and β-tectorin.29 Otoancorin mediates the attachment of the tectorial membrane to the apical surface of the HCs (Fig. 5–3). TECTA encodes α-tectorin, OTOA encodes otoancorin, and COL11A2 encodes the type XI collagen subunit 2. Mutation in any of these three genes results in SNHL, which can be dominant, recessive, NHL, or syndromic HL.29
COCH encodes cochlin, an extracellular matrix protein that is abundantly expressed throughout the inner ear, except sensory hair cells.50 Mutations in COCH at the DFNA9 locus lead to late-onset, progressive NHL, which is characterized by acidophilic mucopolysaccharide deposits in the cochlea and vestibular organs,51 SNHL, and vestibular symptoms similar to Meniere’s disease.52 Interestingly, only missense mutations of the COCH gene have been identified in association with DFNA9 NSHL.53
Mitochondrial Deafness
Mitochondrial mutations have recently been identified in association with some maternally inherited HLs. These can result from heteroplasmic or homoplasmic states with respect to the mitochondrial DNA pool. Heteroplasmy refers to a mixture (usually two, but “multiplasmy” has been reported) of mitochondrial genotypes, whereas homoplasmy refers to a single mitochondrial genotype present in all of the cells of the body.54 Mitochondrial mutations are associated with some forms of syndromic as well as NHL.
The systemic neuromuscular syndromes MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes), MERRF (mitochondrial encephalomyopathy with ragged red fibers), and Kearns-Sayre syndrome frequently present with hearing loss, among other symptoms.55 They are caused by heteroplasmic mitochondrial DNA mutations that manifest in nerves and muscle tissue, where energy requirements are highest, and, therefore, mitochondria are more abundant and active. HL results from generalized neuronal dysfunction.56
A form of inherited SNHL associated with diabetes mellitus results from several distinct heteroplasmic mutations in the mitochondrial transfer RNA (tRNA) genes. In these patients, HL develops after diabetes, but is of early onset with a severe phenotype. Interestingly, these include the A3243G mutation within the mitochondrial gene encoding tRNAleu(UUR), which is the same mutation that results in MELAS syndrome.56
A mitochondrial genome 1555A→G mutation within the gene encoding 12S ribosomal RNA is associated with aminoglycoside susceptibility to HL. Additionally, a progressive high-frequency SNHL and permanent tinnitus, even in the absence of exposure to aminoglycosides, may occur in individuals harboring this mutation, which is, thus, a form of NHL.57 HL presumably results from increased susceptibility to cochlear injury in response to various environmental factors.
Presbycusis is thought to be inherited as a mitochondrial mutation, but the specific mutation(s) have yet to be identified. It is thought to involve at least one of the genes for the oxidative phosphorylation pathway, and the mitochondrial cytochrome oxidase II gene is a likely candidate.54,58
Besides the particular mutation in a single gene, other factors may modify the observed clinical phenotype. Such factors may include the genetic background, modifying genes, and environmental factors, such as noise exposure and ototoxicity. Modifying genes can influence the phenotype resulting from a given mutation at another locus. An extreme example is DFNM1, which acts as a dominant suppressor of DFNB26.59 Modifier genes may explain why the level of HL can range from mild to profound with identical NHL mutations, as well as the phenotypic variance of identical syndromic and mitochondrial mutations.54
Semicircular Canal Dysplasias
Another class of disorders resulting in SNHL, conductive HL, or mixed HL is semicircular canal dysplasia.60 Although traditional hypotheses have held that semicircular canal dysplasias result from arrest in development during the sixth week of gestation, several cases of semicircular canal dysplasia or aplasia have been reported with normal cochlear development.61,62 The majority are nonsyndromic, although 12.5% were associated with known syndromes in a series of 16 cases.62 The genetic mutations leading to isolated semicircular canal abnormalities have yet to be identified.
 Identification of Deafness Genes Utilizing Transgenic Animal Studies
Identification of Deafness Genes Utilizing Transgenic Animal Studies
The study of mouse genetics has been indispensable in terms of the knowledge we have been able to glean about human development and disorders utilizing mice. Creating homologous mutations in experimental mouse models enables us to determine the function of genes mutated in human disorders. Techniques for developing animal models include creating transgenic animals, insertional mutagenesis, and site-directed mutagenesis.
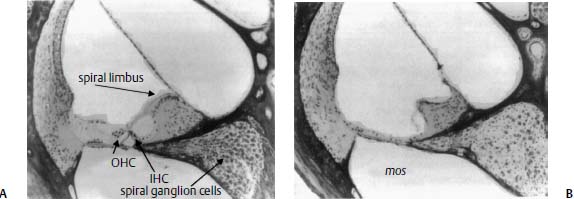
Figure 5–5 Twenty-micrometer sections of the organ of Corti of a transgenic animal overexpressing mos. Compared with normal (A), note the total loss of hair cells, supporting cells, and pillar cells in the mos organ of Corti (B). The tectorial membrane appears somewhat contracted and the normally convex surface of the spiral limbus is scooped out in a deep trough in the mos cochlea. There is also a marked loss of spiral ganglion cells in Rosenthal’s canal. (Original magnification ×100.) (From Rauch S. Malformation and degeneration in the inner ear of mos transgenic mice. Ann Otol Rhinol Laryngol 1992;101:430–436. Reprinted with permission.)
Transgenic Animals
The insertion of a man-made gene into the genome of a normal mouse embryo can result in the production of a transgenic mouse. The artificial gene is composed of a tissue-specific promoter sequence linked to the sequence coding for the protein of interest. This construct is then inserted into the fertilized mouse embryo through various means, including physical injection or retroviral transfection. If the transgene inserts into the genomic DNA and if the embryo survives, the adult mouse will pass the gene on to future generations. In any transgenic animal, if there are cells that express a TF that interacts with the promoter, the gene product will be expressed in these cells and may affect their function. Thus, Rauch63 showed that several lines of transgenic mice in which the mos
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


