Chapter 22 Hurthle Cell Tumors of the Thyroid
 This chapter contains additional online-only content, available on expertconsult.com.
This chapter contains additional online-only content, available on expertconsult.com.
Introduction
In 2011, the there were approximately 48,000 new thyroid cancer cases in the United States: 36,500 in females and 11,500 in males.1 Hurthle cell carcinoma (HCC) of the thyroid gland is an uncommon tumor, accounting for 3% to 10% of all thyroid cancers, and therefore few institutions have significant experience with this condition. Because of its rarity, the natural history and optimal management of patients with HCC remains a subject of much debate. The clinical course of these tumors is unpredictable with reports of apparently benign Hurthle cell adenomas (HCAs) that later behave in a malignant manner and metastasize.2,3 Further, the often aggressive behavior of Hurthle cell tumors (HCTs) has led many to advocate aggressive surgical and adjuvant treatments. However, there has been progress over the years, which has led to an updated classification of Hurthle cell tumors, based on molecular marker testing, offering an improved understanding of its biology and the stratification of treatment of this unusual lesion.
Pathology
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Max Askanazy, a German pathologist, was the first to describe the Hurthle cell in 1898. However, these cells are mistakenly named after the German physiologist Hurthle, who actually described the interfollicular C-cells. Hurthle cells, also known as oncocytes or oxyphilic cells, are seen in both non-neoplastic (Hashimoto thyroiditis, nodular, and toxic goiter) and neoplastic conditions of the thyroid gland (see Chapter 44, Surgical Pathology of the Thyroid Gland). Oncocytes are large polygonal cells characterized by abundant intensely eosinophilic granular cytoplasm on hematoxylin-eosin staining as a result of accumulation of mitochondria. They have a large pleomorphic hyperchromatic nucleus with a prominent nucleolus. Controversy exists about the origin of Hurthle cells in the thyroid gland, although they are generally thought to be derived from the follicular epithelium. Oncocytes are believed to develop as a result of metaplasia that occurs in inflammation and other situations that are associated with cellular stress. A proliferation of oncocytes gives rise to hyperplastic and neoplastic nodules. Hurthle cells can also be seen in tissues outside of the thyroid gland, including the pituitary gland, salivary gland, pharynx, esophagus, trachea, larynx, kidney, and liver.
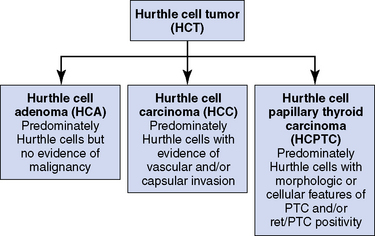
By definition, Hurthle cell neoplasms are encapsulated lesions composed mainly of Hurthle cells with a predominance greater than 75% of the cell population. Other names used for these neoplasms are oxyphil tumor and oncocytoma. There are few to no lymphocytes, which are typically and prominently seen in conditions like Hashimoto’s thyroiditis where Hurthle cells may also be present. Distinguishing a benign neoplasm from a malignant cancer based on cytologic analysis of fine-needle aspiration biopsy is not possible. Definitive differentiation of HCC from an HCA is based on the presence of vascular or capsular invasion on histologic sections or evidence of extrathyroidal spread and local nodal or distant metastases. The reported incidence of malignancy in a Hurthle cell neoplasm is highly variable, partly because of differing histologic interpretations in segregating benign and malignant lesions; overall about 33% demonstrate obvious features of a malignant lesion.4 This may be a major factor in the wide-ranging mortality rates reported in different studies, which have caused much of the controversy regarding the natural behavior and history of HCTs.
 Please see the Expert Consult website for more discussion of this topic, including Figures 22-1 to 22-5.
Please see the Expert Consult website for more discussion of this topic, including Figures 22-1 to 22-5.
In 1988, the World Health Organization classified Hurthle cell carcinoma as a subtype of follicular thyroid carcinoma, oxyphilic type. However, it should be noted that papillary thyroid carcinomas (PTCs) composed of Hurthle cells (oncocytic PTCs) have been also been described (see Chapter 44, Surgical Pathology of the Thyroid Gland). The classification of these PTC lesions is based on the presence of papillary architecture and not on the characteristic nuclear features of PTC. In tumors lacking papillary architecture, the recognition of the diagnostic nuclear features of PTCs is made more difficult by the nuclear hyperchromasia seen in the Hurthle cells and can lead to diagnostic uncertainty. Based on more recent molecular studies, it is now believed that a subset of oncocytic thyroid tumors represents a variant of well-differentiated papillary thyroid cancer (see Chapters 18, Papillary Thyroid Cancer, 19, Papillary Thyroid Microcarcinoma, and 44, Surgical Pathology of the Thyroid Gland). An updated classification of these lesions has been proposed, which offers better clinical prognostication and treatment planning (see Figures 22-1 to 22-5: histologic images of the three different Hurthle cell tumors: adenoma, follicular carcinoma, and papillary carcinoma variants, and see Figure 22-6).5
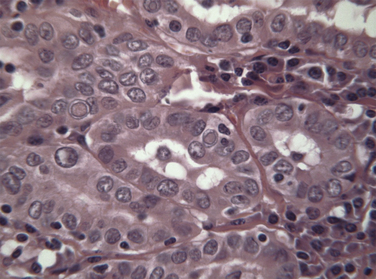
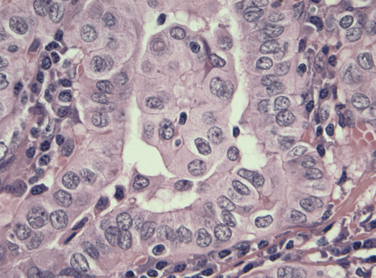
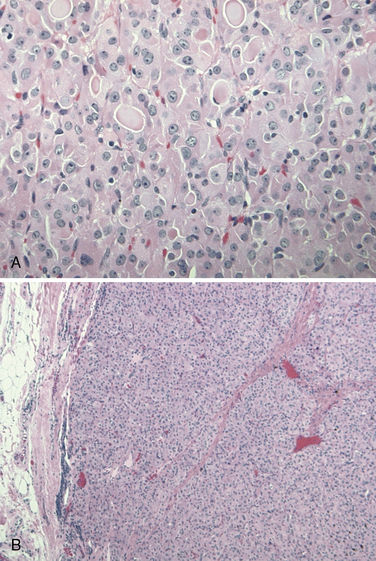
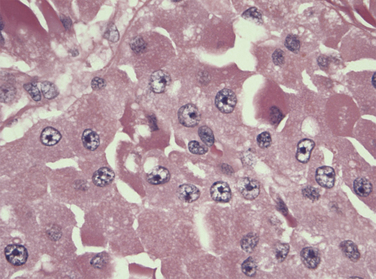
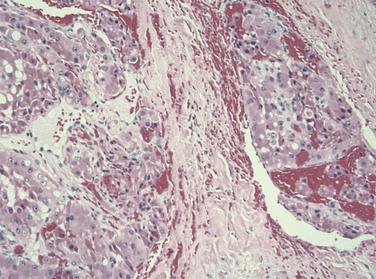
Presentation and Natural History
Most patients with HCC present with a thyroid nodule or mass. The mean age of presentation is between 50 and 60 years of age, approximately 10 years older than the mean age associated with other types of differentiated thyroid cancers.6,7 Several studies have reported a 3:1 preponderance in females.7
Many have traditionally believed that HCCs behave in a more aggressive fashion than other well-differentiated thyroid cancers. However, the reported mortality and survival rates for HCCs are highly variable. Such controversy may be caused by the small sample size, selection biases, and institutional treatment biases. Many earlier studies have reported higher mortality for Hurthle cell carcinoma than for both papillary and follicular thyroid carcinoma, leading many to believe that HCCs are generally aggressive tumors and with a worse prognosis than other well-differentiate thyroid cancers.8–11 In a study of 33 patients treated over 25 years at University of California at San Francisco, Kushchayeva et al. reported a disease-specific survival (DSS) of 74% and 49% at 5 and 10 years, respectively, and a disease-free survival (DFS) of 65% and 40% at 5 and 10 years.8 The authors also reported a metastatic rate of 36% with a recurrence rate of 24% in these patients. Lopez-Penabad et al. reported a similarly high mortality rate of 40% associated with HCC in their cohort of 89 patients managed at the MD Anderson Cancer Center over a period of 50 years.9 These outcome values are in contrast with the 2% overall mortality rate for well-differentiated cancer. Other studies highlight a propensity to regional and distant metastatic behavior of HCCs. In a review of 59 patients with HCC treated over 60 years, Stojadinovic et al. reported a 33% rate of distant metastases and a 21% rate of nodal metastasis.10 Ruegemer et al. also demonstrated a 34% rate in distant metastases in HCCs as compared to only 7% to 19% rate in papillary thyroid cancer (PTC) and follicular thyroid cancers (FTCs), respectively.11
However, more recent studies and larger public database surveys have suggested better survival figures for HCCs, comparable to that of other differentiated thyroid cancer types.7,12,13 Our group reviewed 45 HCCs and demonstrated a favorable overall outcome (DSS 96% at 5 years) despite a high incidence of regional cervical metastases (55%).12 The high incidence of lymphatic spread and the relatively benign clinical course observed in this study is more in keeping with the clinical behavior of the well-differentiated PTCs. Bhattacharyya examined the Surveillance, Epidemiology and End Results database (SEER) for the10-year period between 1988 and 1998 for Hurthle cell carcinoma and identified 555 cases of nonmetastatic HCCs. He reported an overall mean 5-year and 10-year survival of 85% and 71%, respectively.7 Mean survival figures for 411 matched FTCs were comparable. The author reported age and tumor size as the only variables to be predictors of poorer outcome on multivariate analysis.7
Recent work on molecular testing for ret/PTC rearrangements has resulted in a subclassification of some HCTs into a papillary variant of HCC, called Hurthle cell papillary thyroid carcinoma (HCPTC), has been described (Figure 22-6).4,14 These lesions have been shown to display a more favorable clinical behavior compared to nonpapillary HCCs. A recent study, in 2006, of 42 HCPTCs reported an overall 5-year and 10-year survival of 94% and 87%, respectively, and the 5-year and 10-year disease-free survival of 93% and 81%, respectively. This study described a high lymph node metastasis rate of 31% (13/42) and distant metastases in only 5% (2/42) of patients. Locoregional occurrence was seen in 10% (4/42) of patients with 7% (3/42) dying from their disease.15 HCPTCs appear to behave in a similar fashion to their papillary thyroid cancer counterparts and are associated with good prognosis despite a high incidence of nodal metastases at diagnosis. This is in contrast to the nonpapillary HCCs that may represent a much more aggressive disease associated with higher distant metastases and a significantly poorer survival outcome. The segregation of these HCPTCs from the larger group of more aggressive nonpapillary HCT is possible through molecular testing (see the subsequent “Tumor Classification by Ret/PTC Genetic Testing” section).
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.Pathogenesis
As yet, there is no clear association between HCCs and environmental or infective carcinogens. As with other solid tumors, it is widely believed that progressive transformation of normal cells toward increasingly more malignant cell types through a stepwise accumulation of somatic mutations of genes underlies the pathogenesis of Hurthle cell tumors (see Chapter 17, Molecular Pathogenesis of Thyroid Neoplasia). Oncogenic activation by gene mutations and chromosomal translocations has been shown to be common in thyroid cancers. The Ras oncogene has been shown to be one of the most prevalent oncogenes to be activated in well-differentiated follicular thyroid cancers including Hurthle cell carcinomas.
Ret/PTC Rearrangements
Several groups have recently investigated the ret/PTC rearrangements defining a subset of HCTs as HCPTC.4,14 One group examined 27 HCCs, 4 oxyphil FTCs, 5 oxyphil PTCs, and 16 oxyphil adenomas using RT-PCR for the presence of ret/PTC rearrangements and demonstrated 4 of 5 oxyphil PTC and 0 of 4 oxyphil FTC to be ret/PTC positive. Interestingly, 7 of 27 HCC and 3 of 16 oxyphil adenomas were also positive for the ret/PTC aberration.17 Their data describe a similar percentage of ret/PTC positivity in HCCs as seen in classical PTCs. In addition, Chiappetta et al. examined 49 Hurthle cell lesions (20 adenomas, 19 carcinomas, and 10 hyperplastic lesions) and demonstrated 12 of 20 (60%) ret activation in adenomas, 9 of 19 (47%) in carcinomas by immunohistochemistry and ret/PTC1 positivity using RT-PCR in 7 of 12 (58%) adenomas and 4 of 7 (57%) carcinomas. No ret/PTC positivity was seen in normal thyroid tissues or benign hyperplastic Hurthle lesions.18
Taken together, these studies suggest a papillary lineage in many HCCs and the potentially critical role of ret/PTC mutation (and other genetic abnormalities) in the development of this subset Hurthle cell tumors.4,17 Further, given the similar rate of ret/PTC positivity in many HCAs and HCCs, it has been suggested that HCTs that harbor these gene abnormalities should be considered malignant despite the lack of histologic findings typically associated with malignancy in these HCAs. This hypothesis would help to explain how apparently benign histologic lesions may give rise to distant metastases later.1,2 In many earlier studies, prior to the advent of molecular testing, it may be that lesions identified as HCCs may have been only the most aggressive forms, easily identifiable histologically, with many true malignant lesions being incorrectly diagnosed as benign on histologic evaluation. Clearly the findings from the preceding studies support the diagnostic role of genetic testing in HCTs for improved tumor classification, clinical prognostication, and thus better treatment stratification (Figure 22-7).
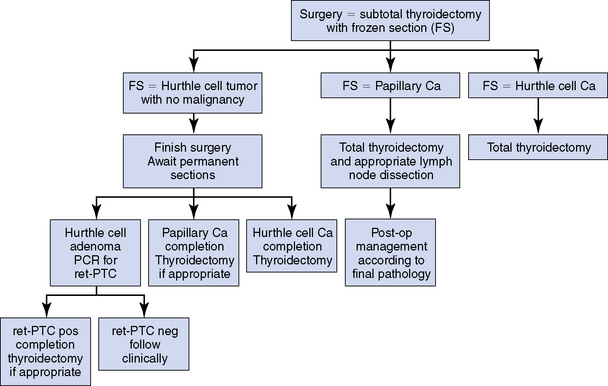
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Both BRAF and ret/PTC mutations are common genetic aberrations found in papillary thyroid cancers (see Chapter 17, Molecular Pathogenesis of Thyroid Neoplasia). Further, the BRAF V600E mutation is found almost exclusively in ret/PTC negative tumors. One group examined BRAF abnormalities and showed no mutations in 44 well-characterized oncocytic follicular thyroid tumors, further supporting the concept that such oncocytic follicular neoplasms are likely more closely related to follicular than papillary cancer lineage.19
Other molecular testing methods, such as DNA-ploidy measurements,31 have been suggested to provide prognostic information. However, a lack of consistency and reproducibility of these findings has precluded their use in routine clinical practice. Mitochondrion-related genetic abnormalities, such as mutations in mitochondrial DNA, have been described in HCTs.20,21 In one study almost all Hurthle cell tumors displayed a point mutation in mitochondrial DNA.22 Hurthle cell neoplasms are often aneuploid, and DNA-ploidy studies have demonstrated a high frequency of abnormal flow cytometry profiles.23 Abnormal DNA content on flow cytometry has been shown to be a potential marker for more aggressive clinical behavior but shows interestingly no ability in differentiating benign from malignant tumors. Other molecular targets have been investigated for their role in the development and prognostication of HCTs. These include mutation in the GRIM gene (a mitochondrial gene),20 p27 expression, and LOH studies.24 However, no consistent and conclusive evidence have been demonstrated as to their etiologic role.



