22
Hereditary Hearing Impairment
Daniel I. Choo and John H. Greinwald Jr.
Although many areas of medicine have benefited from the exponentially expanding knowledge base of molecular genetics and the underlying molecular biology of human diseases, few branches of medicine have been as dramatically affected as clinical otology and the diagnosis and management of patients with hereditary hearing impairment (HHI). Over the course of just one decade, genes responsible for some of the most common forms of HHI have been identified, and “routine” laboratory assays to identify mutations of these genes have become almost uniformly available in most major medical centers across the United States. These advances in the molecular genetics of hearing loss have translated into a markedly improved ability of clinicians to precisely diagnose the cause of hearing loss in a large population of patients in whom all traditional diagnostic testing would previously have been uninformative.
Given the fact that hearing impairment is the most common sensory deficit in the United States (affecting 2 to 3 per 1000 live births) and affects about 28 million Americans, this improved diagnostic ability has tremendous clinical relevance. Furthermore, as many as 5% of children have a demonstrable hearing loss by 18 years of age, with that incidence reaching 40 to 50% by age 75. Taking into account the aging demographic of the United States population, it becomes apparent that more accurate, efficient, and cost-effective diagnosis and management of patients with hearing loss are requisite. This chapter illustrates how the emerging era of molecular medicine translates into the field of clinical otology, particularly the care of patients with HHI (see also Chapter 5).
 Basic Principles and Nomenclature in Human Genetics
Basic Principles and Nomenclature in Human Genetics
To meaningfully discuss the genetic mechanisms of HHI, a grasp of some basic terminology is required. The core material composing the human genome is linear strands of deoxyribonucleic acid (DNA) whose precise sequence of base pairs creates genes that are organized into 23 pairs of chromosomes: 22 autosomes and 1 pair of sex chromosomes (in total, 46 chromosomes). An X and a Y chromosome for males and 2 X chromosomes for females determine sex.
Genes have been further dissected to identify regions within their DNA sequence that have specific functions. In general, each gene contains (1) DNA that has the actual coding sequence(s) (called exons) that contain the information for synthesizing proteins; (2) interspersed (noncoding) DNA between these exons (called introns); and (3) regulatory regions of DNA before and after the exons that are referred to as upstream or downstream regions, respectively. Particularly relevant for most genes are upstream regions termed promoters that bind regulatory proteins and determine when genes will be actively read or transcribed and made into messenger RNA (mRNA). mRNA is then translated or made into protein by cells to carry out the actual function of that gene.
Mutations are abnormal alterations of the DNA sequence in genes that then impact their ability to guide cellular processes. Nonsense mutations, for example, result in an early “stop” in the transcription or reading of the DNA. No mRNA or protein is synthesized. In contrast, missense mutations result in an abnormal transcript or protein that is synthesized but abnormal because of the mutation. In this case, the abnormal protein may not function normally and contribute to the phenotype.
The other relevant terminology can best be explained by discussing a specific example of genetic hearing impairment: gap junction β2 (GJB2)-related HHI. (GJB2-related hearing loss is discussed in greater detail later in this chapter.) GJB2 refers to the specific gene located on chromosome 13 at a locus (or position on the chromosome) designated 13q11; “13” refers to the chromosome number, with “q11” further specifying a position along the long arm of chromosome 13. Every person carries two copies of the GJB2 gene in every cell of the body. One copy (or allele) of the GJB2 gene is inherited from the biologic mother whereas the other allele is inherited from the biologic father. Several variants or forms of the GJB2 gene (and most other genes) exist. If both alleles of the GJB2 gene are identical, then the individual is homozygous at the GJB2 locus. If the alleles are different, then the individual is heterozygous for GJB2. This description of an individual’s genetic composition is termed the genotype for any given trait. In distinction, the physical trait(s) that a person displays (in this case, hearing impairment) is termed the phenotype. In the case of HHI, the onset and nature of the hearing impairment (i.e., the phenotype) can be largely determined by the patient’s genotype (i.e., which alleles of the GJB2 gene a person carries).
Alleles also can function in a dominant or recessive manner depending on whether the presence of a single allele can determine/produce a phenotype (dominant) or whether two copies of an allele are required to produce a phenotype (recessive). In most cases of GJB2-related hearing impairment, both copies of a person’s GJB2 gene need to be abnormal to cause a hearing impairment (i.e., autosomal recessive). In other autosomal dominant processes, one abnormal copy of the gene (even in the presence of one normal allele) is sufficient to produce a phenotype (e.g., some forms of Waardenburg’s syndrome caused by MITF mutations). Dominant traits typically do not skip generations, and there is a 50% chance that a heterozygous parent will transmit the abnormal allele to offspring. In contrast, recessive traits do not typically appear in each generation, and there is a 25% chance that two heterozygous parents would produce an affected child.
One feature that can affect this simple inheritance concept is the phenomenon of penetrance (or ability of the gene to manifest the phenotypic traits). In some autosomal dominant disorders, a patient may carry an abnormal gene but not display any physical abnormalities (i.e., incomplete penetrance). Furthermore, dominant disorders also can show variable expressivity, whereby different family members who carry the same abnormal gene can present with different manifestations of the abnormality. In the case of HHI, this might present as varying degrees of hearing impairment in family members all carrying the same genetic abnormality.
Sex-linked or X-linked traits demonstrate a different inheritance pattern due to males carrying only one X chromosome. For example, an X-linked recessive trait can lack phenotypic expression if carried in a female who is heterozygous. However, male offspring of this female would have a 50% chance of inheriting the gene, and expression of the gene would occur in a male child because of the lack of a second X chromosome. The female offspring of an X-linked female carrier have a 50% risk of carrying such a trait. But because males inherit their father’s Y chromosome (i.e., not the X-linked recessive allele), they are unaffected and do not become carriers. In contrast, females inherit their father’s X-linked allele and necessarily become carriers of the allele.
There are circumstances in which diseases or phenotypes are caused by abnormalities that involve all or portions of chromosomes (as opposed to individual genes as described above). Most of these chromosomal abnormalities result in clinical conditions with developmental delays and a spectrum of congenital anomalies, the exception being instances in which sex chromosomes are affected. As an example, trisomy 21 (Down syndrome) is caused by the presence of a third copy of an entire chromosome (chromosome 21). Individuals with extra X or Y chromosomes tend to have milder phenotypes such as Klinefelter syndrome (47, XXY or XYY) or Turner syndrome (45 X).
Remarkable advances in the knowledge base of molecular biology and human genetics of normal and impaired hearing have been made over the past several years. However, greater detail into these topics is beyond the scope of this chapter. For further reading and more in-depth review of these areas, the reader is referred to resources such as the Hereditary Hearing Loss Homepage (www.webhost.ua.ac.be/hhh).
 Autosomal Recessive Disorders of Hearing
Autosomal Recessive Disorders of Hearing
Overall, most genetic hearing disorders are transmitted in an autosomal recessive mode of inheritance. Furthermore, for those hearing impairments presenting in childhood, roughly 80% are inherited recessively. In the vast majority of patients (80–90%), the HHI is nonsyndromic (i.e., with no other abnormalities/phenotypes that can be identified). From a clinical perspective, this poses significant challenges in the workup and evaluation of an otherwise healthy-appearing child presenting with sensorineural hearing impairment (SNHI). Although most children do not have any other anomalies, a thorough evaluation is necessary to identify the 10 to 20% of patients who have other features that would point toward a syndromic etiology and potentially a different evaluation and management pathway. A very common scenario is to identify an isolated child with SNHI in a family with no known history of HHI. The medical evaluation of such a patient requires methodical history taking and physical examination (in addition to ancillary testing, such as genetic testing) to differentiate nongenetic causes of hearing impairment from those that are inherited in an autosomal recessive fashion. The sections below cover the different categories of autosomal recessive, autosomal dominant, sex-linked, mitochondrially related and gross chromosomal abnormality-related HHI. To further structure the discussion, each category is then subdivided into syndromic and nonsyndromic forms of HHI where appropriate.
Autosomal Recessive Nonsyndromic Hearing Impairment (ARNSHI)
The rapidly advancing field of auditory molecular genetics has shown that ARNSHI is a genetically heterogeneous disorder, meaning that many genetic causes have been identified that can cause HHI. As an illustration, 41 loci for ARNSHI have already been reported. The nomenclature for these genetic loci uses the prefix DFNB, with DFN indicating deafness and B indicating an autosomal recessive mode of inheritance. (In contrast, the prefix DFNA would be used for deafness transmitted in an autosomal dominant fashion A, discussed below). To date, 22 genes have been identified as being involved in ARNSHI (Table 22–1) with DFNB1 (the first autosomal recessive disorder) responsible for 30 to 40% of all cases of ARNSHI.
DFNB1
The gene identified at the DFNB1 locus is GJB2, which encodes for a protein called connexin 26.1–4 Connexin 26 represents one of a family of gap junction proteins that form intercellular channels that allow low molecular weight molecules to travel from cell to cell. In the inner ear, connexin 26 is one of several gap junction proteins expressed in the epithelia lining the cochlea and has been reported to be involved in potassium recycling that is critical to hair cell transduction of sound stimuli into electrical signals.
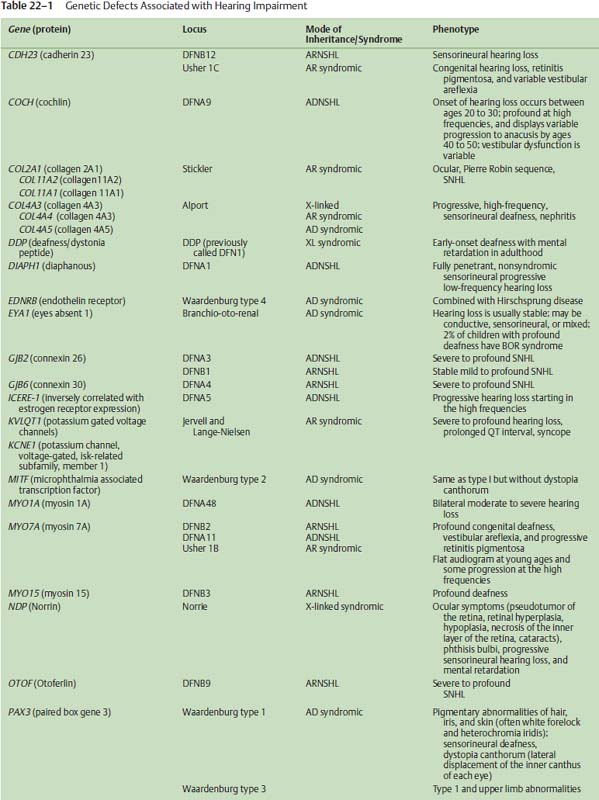
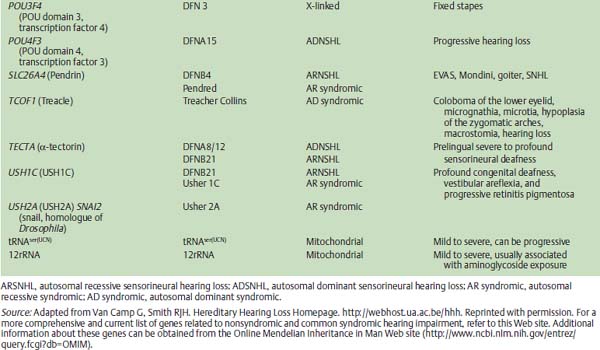
Although GJB2-related HHI has been identified in almost all regions of the world, certain ethnic backgrounds and populations display different rates of specific GJB2 mutations. For example, deletion of a guanine residue at nucleotide position 35 (35delG) is one of the most common mutations found in patients with ARNSHI and is particularly common in those of European descent. In contrast, a deletion of a thymine at position 167 (167delT) is much more common in Ashkenazi Jewish populations, whereas a 235delC mutation is more frequently identified in Asian patients.
Overall, DFNB1 is believed to cause 20% of all childhood HHI and among those children with more severe degrees of hearing impairment (e.g., thresholds >70 dB), almost 40% are due to DFNB1. Examining those pediatric patients with milder forms of hearing impairment, the rate of GJB2 mutations drops to a smaller but nonetheless significant rate of 10 to 15%. Further illustrating the clinical relevance of DFNB1 is the estimated frequency of heterozygous carriers of deafnesscausing GJB2 mutations in the Caucasian U.S. population of 1 in 40. Taking together the high frequency of mutations in GJB2 and the relatively small size of this gene, these factors make genetic screening and molecular diagnosis of GJB2-related hearing loss extremely feasible and attractive.
Related to GJB2 HHI, mutations of GJB6 also have been found in association with GJB2 mutation, termed a double heterozygous state that may be another deafness-causing genotype. A large deletion of the GJB6 upstream region has been found by itself to produce a severe hearing impairment phenotype.5 However, the greater significance of GJB6 mutations may lie in the fact that as many as 10% of patients with ARNSHI and GJB2 mutation also carry a large deletion of GJB6. Also, the colocalization of GJB2 and GJB6 to the same region of chromosome 13 may indicate these genes share a common regulatory region (i.e., a locus control region). Accordingly, gap junction gene mutations have an obvious importance in HHI, and although the basic molecular and cellular mechanisms of gap junction-related hearing impairment remain somewhat nebulous, this initial ability to molecularly diagnose patients with specific genetic mutations responsible for their HHI presents a clinically useful and necessary first step in translating molecular genetics to the patient care arena.
Recently published data demonstrate how these molecular data can be incorporated into clinical practice to improve efficient diagnosis and cost-effectiveness.6 For pediatric patients with ARNSHI and moderately severe or worse hearing thresholds (e.g., >60 dB), GJB2 testing is recommended as the first line of evaluation. If hearing thresholds are better than 60 dB, GJB2 testing should be considered after temporal bone computed tomography (CT) has been performed. For these patients with normal temporal bone CT scans, GJB2 testing is then appropriate. If the CT scan is abnormal, then genetic testing for SLC26A4 (Pendred gene) mutations should be obtained.
Interestingly, GJB2 mutation data have now been analyzed to correlate specific mutations with degrees of hearing impairment.7 As an example, patients with two nonsense mutations (biallelic nonsense mutations, e.g., 35delG/35delG) have an extremely high likelihood of ultimately demonstrating a bilateral severe to profound hearing impairment. In contrast, patients with two missense mutations (e.g., M34T/M34T) are much more likely to demonstrate mild to moderate hearing impairments. Such prognostic factors have gained tremendous relevance since the implementation of universal newborn hearing screening programs across the United States. As infants are identified at birth after not passing their newborn hearing screening, clinicians will be challenged with determining management strategies as early as possible to maximize the infant’s hearing and language skill development and to take advantage of early windows of opportunity, including cochlear implantation at 12 months of age for those infants with more severe degrees of hearing impairment. By obtaining GJB2 genotyping shortly after confirming a hearing impairment, clinicians can assimilate these molecular genetic data and more accurately identify those infants who are likely to demonstrate severe-to-profound hearing impairments and may require different and earlier interventions to let them reach their full communicational potential. For example, if auditory neuropathy-dyssynchrony occurs at the level of the spinal ganglion, cochlear implantation results still may be surprisingly good.
Otoferlin
One other noteworthy form of ARNSHI is HHI due to mutations of the Otoferlin (OTOF) gene.8,9 OTOF mutations have been associated with a specific form of hearing impairment known as auditory neuropathy or auditory dyssynchrony (AD). AD is audiometrically characterized by an abnormal auditory brainstem response (ABR) with limited waveforms other than the cochlear microphonic. Additionally, patients with AD often demonstrate present otoacoustic emissions (OAEs), suggesting intact cochlear outer hair cell function. In these patients with AD, behavioral hearing thresholds can range from mild to profound hearing loss. Estimates indicate that 5 to 10% of patients with SNHI have AD. The optimal management of these patients is challenging, as some do not benefit from traditional amplification or other assistive listening devices. Through an improved understanding of the molecular genetics and biology of AD, improved therapies may become apparent.
Autosomal Recessive Syndromic Hearing Impairment
Jervell and Lange-Nielsen Syndrome
Jervell and Lange-Nielsen (JLN) syndrome represents a rare but very significant form of syndromic HHI. In JLN, infants are typically identified with a severe-to-profound congenital hearing impairment. The critical comorbidity is a prolongation of the QT interval on electrocardiography that can cause episodes of syncope or sudden cardiac death. If properly identified after presentation with a congenital hearing impairment, these infants can be managed with beta-blockers (e.g., propranolol), which are effective in controlling the cardiac condition. Accordingly, an astute clinician can detect a potentially life-threatening cardiac condition in infants by obtaining an electrocardiogram on infants with severe-to-profound SNHI. Other patients with SNHI and unexplained seizures or a family history of syncope or sudden death should similarly be sent for an electrocardiogram if not a full cardiologic evaluation.
Genetically, some cases of JLN can be attributed to mutations of a potassium channel gene, KVLQT1.10 Other mutations of this gene have been identified but are associated with isolated cardiac conduction defects and not SNHI. Mutations of KCNE1 have also been identified in some families with JLN,11 indicating again that the causes of ARNSHI can be heterogeneous.
Pendred Syndrome and Enlarged Vestibular Aqueduct Syndrome
Although the association of goiter and SNHI has been known since the late 1800s, the molecular basis for Pendred syndrome (euthyroid goiter and SNHI)12,13 was not elucidated until much more recently.14–16 The Pendred syndrome gene (PDS) is also referred to as SLC26A4 (solute carrier family 26, member 4) and encodes for the Pendrin protein that functions as an anion transporter. The hearing impairment in Pendred syndrome varies in terms of severity as well as age of onset. Some patients display a profound loss from birth, whereas others show later onset SNHI of lesser severity. Diagnostic imaging studies routinely show cochlear hypoplasias (Mondini-type deformities) in concert with enlarged vestibular aqueducts. However, mutations of the PDS gene have been identified in some patients with isolated enlarged vestibular aqueducts on CT imaging (see later in chapter).
Previously, the diagnostic test for Pendred syndrome was an abnormal perchlorate discharge test that identified defects in the organification of iodine that was typical of patients with Pendred syndrome. However, this assay is not specific for Pendred syndrome, and its sensitivity has not been definitively determined. Thyroid function testing for patients with suspected Pendred syndrome is still routinely recommended. It is noteworthy to mention that goiter typically does not manifest until 8 years of age or later, and exogenous thyroid hormone therapy is often the most appropriate therapy. Genetic testing for PDS mutations provides another manner for identifying these patients who often present at a young age with SNHI and CT abnormalities of the inner ear. For families with the full Pendred phenotype (i.e., hearing impairment and euthyroid goiter), there is little evidence of genetic heterogeneity. About 1 to 2% of patients with severe-to-profound hearing impairment can be attributed to PDS mutations.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


