Genetics of Eye Disease
Terri L. Young
Leila M. Khazaeni
J. Bronwyn Bateman
IN 1903, SUTTON noted parallels between chromosome behavior and Mendel’s laws, thus identifying genes with chromosomes and marking the beginning of genetics as a science (1). One hundred years later, the fiftieth anniversary of the publication of the proposed double helical structure of deoxyribonucleic acid (DNA) by Watson and Crick occurred on April 25, 2003 (2). Another monumental undertaking—the Human Genome Project initiative to sequence the entire human genome—was also pronounced as completed in 2003 (www.genome.gov). The Human Genome Project is a joint initiative of the Department of Energy (Washington, DC) and the National Institutes of Health (Bethesda, MD), and was initiated with the major goal of sequencing the DNA that defines the human genome (www.ornl.gov/hgmis/home.shtml). The ultimate goal of the project, the complete sequencing of human DNA, was completed as a first draft both by the government and by the privately funded effort of Celera Genomics (www.celera.com) (now acquired by Quest Diagnostics, Inc., Madison, NJ). The public version of the current sequence is accessible at several web sites including the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) and the genome browser at the University of California at Santa Cruz (http://genome.ucsc.edu). Medicine has benefited immensely from this evolution in scientific discovery. A paradigm shift has occurred in the way diseases are classified, with less emphasis on clinical features as categorical constructs and greater emphasis on gene protein dysfunction due to associated DNA sequence alterations.
Genetics is a universal science that embraces all of biology. Lenz wrote in 1936: “Rules that are obeyed the same way in peas and snapdragons, in flies and butterflies, in mice and rabbits, of course also apply to humans” (3). This has repeatedly played out with the increasing pace of gene discovery in the latter part of the 20th century, and into the 21st century, in large part due to genetic defects found in lower species that were then found to be associatively consistent in humans. Modern genetics has allowed the characterization of mutations that cause congenital human disorders and their comparison to mutations in model organisms. The worm (Caenorhabditis elegans), fruit fly (Drosophila melanogaster), zebrafish, and mouse all serve the medical community with modules of evolutionary conserved ontogenetic mechanisms that aid in establishing developmental and functional models of human disease (4). With the advent of modern molecular genetics, the medical community has acquired a common language and a new paradigm.
The current statistics of ophthalmic genetic disorders is large and growing. Searching the Online Mendelian Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih. gov/omim) for the term “eye” yields more than 1,242 entries (5). winter’s diagnostic London Dysmorphology Database (LDDB) lists under the general term “eyes, globes” more than 2,900 genetic ophthalmic conditions as part of expanded programmatic database named GENEEYE (6) (www.lmdata bases.com/). This includes many single congenital anomalies both genetic and sporadic, including all the corneal dystrophies, macular dystrophies, the scores of different reo-cone dystrophies. Specific defects found in nearly 192 genes are associated with corneal and retinal dystrophies, eye tumors, retinitis pigmentosa, cataracts, and glaucoma (http://www. sph.uth.tmc.edu/Retnet/disease.htm). Characterization of gene mutations in specific eye diseases aids in the identification of abnormal proteins that cause disease, and expanded definitions of pathologic processes.
Gene mutations have now been identified for several ophthalmic and systemic disorders with significant sightthreatening ophthalmic consequences. Ophthalmologists, and in particular pediatric ophthalmologists, are often on the frontline in assessing patients and families with such disorders. Knowledge of clinical and molecular features of the disease—such as developmental age of onset, heritable probability, DNA testing parameters, and customized treatment options based on molecular insights and strategies—is paramount to customized patient care.
DATABASES FOR CLINICAL USE
Much of the information collected and discussed in this chapter will no doubt be dated even at publication because of the rapid evolution of research discoveries in genetics. For this reason, it is recommended that inquiries regarding specific clinical entities be made consulting online databases that are updated more quickly than any textbook
chapter. As mentioned above, the OMIM database is without question the reference of choice of geneticists and genetic counselors for information regarding syndromic and nonsyndromic clinical entities for which a genetic basis has been discovered. OMIM’s delay of comprehensive coverage of the scientific literature is less than 2 weeks. The LDDB is structured by the symptoms, anatomical sites, or tissues involved. Many genetic and clinical centers also use the Australian Pictures of Standard Syndromes and Undiagnosed Malformation (POSSUM) database at http://www.possum.net.au/, which was launched in March 1987. Similar to LDDB, this program uses a hierarchic trait search list. The syndrome descriptions include the OMIM number, a list of synonyms, pictures of different patients at different ages, clinical and genetic comments, references, and trait lists. The program is continuously updated. The Human Gene Mutation Database (http://www.hgmd.org) at the Institute of Medical Genetics in Cardiff, Wales, curated by Cooper and colleagues since 2000, is at present the most useful general mutation database (7). It covers the scientific literature, references genes (at this writing more than 3,682 genes), and mutations discovered to date of those genes (more than 97,797), with links to approximately 250 open locus-specific databases, such as the retinoblastoma gene RB1 (http://www.d-lohmann.de/Rb/). The web site of the HUGO (Human Genome Organisation) Mutation Database Initiative (8) at (www.genenames.org) contains a number of links to locus-specific, central, general, national, and ethnic mutation databases. Newer database, such as The 1000 Genomes Project Consortium, is an integrated map of genetic variation from 1,092 individual human genomes from 14 populations, constructed using a combination of low-coverage whole-genome and exome sequencing to provide a validated haplotype map of 38 million single nucleotide polymorphisms, 1.4 million short insertions and deletions, and more than 14,000 larger deletions (www.1000genomes.org/).
chapter. As mentioned above, the OMIM database is without question the reference of choice of geneticists and genetic counselors for information regarding syndromic and nonsyndromic clinical entities for which a genetic basis has been discovered. OMIM’s delay of comprehensive coverage of the scientific literature is less than 2 weeks. The LDDB is structured by the symptoms, anatomical sites, or tissues involved. Many genetic and clinical centers also use the Australian Pictures of Standard Syndromes and Undiagnosed Malformation (POSSUM) database at http://www.possum.net.au/, which was launched in March 1987. Similar to LDDB, this program uses a hierarchic trait search list. The syndrome descriptions include the OMIM number, a list of synonyms, pictures of different patients at different ages, clinical and genetic comments, references, and trait lists. The program is continuously updated. The Human Gene Mutation Database (http://www.hgmd.org) at the Institute of Medical Genetics in Cardiff, Wales, curated by Cooper and colleagues since 2000, is at present the most useful general mutation database (7). It covers the scientific literature, references genes (at this writing more than 3,682 genes), and mutations discovered to date of those genes (more than 97,797), with links to approximately 250 open locus-specific databases, such as the retinoblastoma gene RB1 (http://www.d-lohmann.de/Rb/). The web site of the HUGO (Human Genome Organisation) Mutation Database Initiative (8) at (www.genenames.org) contains a number of links to locus-specific, central, general, national, and ethnic mutation databases. Newer database, such as The 1000 Genomes Project Consortium, is an integrated map of genetic variation from 1,092 individual human genomes from 14 populations, constructed using a combination of low-coverage whole-genome and exome sequencing to provide a validated haplotype map of 38 million single nucleotide polymorphisms, 1.4 million short insertions and deletions, and more than 14,000 larger deletions (www.1000genomes.org/).
BASIC GENETICS CONCEPTS
The eye is affected relatively early in the course of many genetic metabolic diseases; for some disorders, the ocular manifestations are unique and diagnostic. The eye is a complex organ with unique and specialized structures and biochemical functions related to vision. For these reasons, it is particularly vulnerable to genetic mishaps and inborn errors of metabolism.
The hereditary bases of diseases that affect the eye include several broad categories: single-gene mutations consistent with Mendelian inheritance patterns, chromosomal aberrations, cytoplasmic mitochondrial inheritance, and multifactorial inheritance; monogenic disorders can be divided into those that affect only the eye and ocular adnexa, and those that affect other systems in addition to the eye.
The term phenotype is defined as “the entire physical, biochemical, and physiological nature of an individual, as determined by his genetic constitution and the environment in which he develops; or, in a more limited sense, the expression of some particular gene or genes, as classified in some specific way” (9), and usually refers to either a physical feature or features of the individual or the biochemical assay of a gene product. It is a measurable physical or biochemical parameter that is determined by the interaction of the genetic constitution (genotype) with the environment. A wild-type phenotype is considered the normal or standard clinical (physical or biochemical) feature. The genotype may refer to the sum total of an individual’s hereditary material (genome) or it may specify a single gene or gene pair.
At the biochemical level, the basic genetic unit for the orchestration of cellular function and transmission of traits from one generation to the next is a macromolecule consisting of DNA; it is self-reproducing and determines the composition of amino acids to form proteins. With rare exceptions, each cell of an organism contains the same DNA as every other cell, and this material encodes the information for the synthesis of proteins that catalyze enzymatic reactions, function as support structures, regulate intra- and intercellular functions, and determine the fate of a cell. The gene is a sequence of DNA that encodes for a single, specific protein or regulates the expression of a gene; just as the DNA is arranged like beads on a necklace, the genes are similarly aligned. The information contained in the DNA is transcribed to ribonucleic acid (RNA), an intermediary template, which is in turn translated to the protein (Fig. 1.1). The four types of RNA that have been identified are messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), and heterogeneous RNA (hnRNA). mRNA, which forms the template for protein synthesis in the cytoplasm, is formed in the nucleus of the cell from the DNA; tRNA transfers amino acids from the cytoplasm to the specific positions along the mRNA template. The functions of rRNA and hnRNA are less well understood. rRNA is associated with protein in the ribosomes and carries limited genetic information; hnRNA is an intranuclear RNA that may play a regulatory role. In eukaryotic cells (those with a nucleus and nuclear membrane, including human, plant, and protozoan cells but not bacteria), the DNA segments encoding a protein are interspersed with DNA that does not code for that protein; the coding portions are called exons and the noncoding portions introns. During the processing steps, the introns are removed and the exons are fused to form the mature mRNA, which is translated into the protein.
Both DNA and RNA are arranged in a linear fashion and consist of nucleotides, each of which has a base, pentose
(five-carbon sugar—deoxyribose in the case of DNA and ribose in the case of RNA), and phosphoric acid (Fig. 1.2). The bases are either purines (adenine or guanine) or pyrimidines (cytosine or thymine in DNA, and cytosine or uracil in RNA). The pentose sugar and the phosphoric acid form the macromolecular support. The precise order of the bases within the exons of a gene determines the amino acid sequence of the protein for which it encodes. For each amino acid, a triplet of DNA bases (codon) provides the necessary information for the identification of the amino acid in the protein; many amino acids have several codons that encode for them. For example, the amino acid phenylalanine is encoded by the triplet base sequence uracil-uracil-uracil or uracil-uracil-cytosine.
(five-carbon sugar—deoxyribose in the case of DNA and ribose in the case of RNA), and phosphoric acid (Fig. 1.2). The bases are either purines (adenine or guanine) or pyrimidines (cytosine or thymine in DNA, and cytosine or uracil in RNA). The pentose sugar and the phosphoric acid form the macromolecular support. The precise order of the bases within the exons of a gene determines the amino acid sequence of the protein for which it encodes. For each amino acid, a triplet of DNA bases (codon) provides the necessary information for the identification of the amino acid in the protein; many amino acids have several codons that encode for them. For example, the amino acid phenylalanine is encoded by the triplet base sequence uracil-uracil-uracil or uracil-uracil-cytosine.
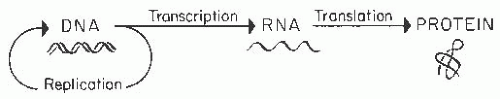 FIGURE 1.1. Relationship between transcription, translation, and replication. |
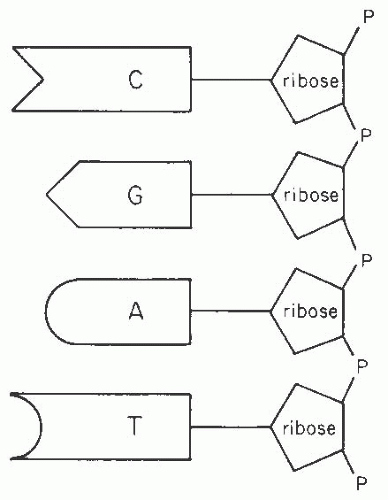 FIGURE 1.2. Nucleic acid structure. |
Genetic disorders may be roughly categorized into those caused by single-gene defects, chromosomal or large DNA abnormalities such as duplications or deletions of large segments, or the effect of more than one gene. For each species, a gene occurs at a particular position (locus) on a specific chromosome. Humans are diploid organisms with two pairs of 22 autosomal chromosomes and two sets of genes—one member of each pair is inherited from each parent. The remaining two chromosomes, X and Y, determine gender. An individual is homozygous for a gene pair if the information specified by each member is identical, and heterozygous if the two encode for different polypeptides. Alternate forms of a gene are called alleles. Some alleles represent common variations not associated with disease (called polymorphisms), others represent disease-producing mutations (alterations of the DNA sequence that cause a change in amino acid sequence which alters the actual protein translated), and others are advantageous to the host under certain conditions. For example, the blood serotypes A, B, and O are common alleles; a normal individual may have AA, AO, BO, AB, or BB blood type. As another example, the genes coding for hemoglobin A (normal), S (sickle), and C are alleles, the hemoglobins S and C being mutations; the end products differ from each other by one amino acid in a chain of 146. Hemoglobin A has the amino acid glutamic acid in the position where valine is found in hemoglobin S and lysine in hemoglobin C. The substitution of glutamic acid alters the function of the protein. Allelism is the source of genetic variation in humans.
Most of the DNA is arranged in discrete chromosomes within the nucleus. A small fraction is within the mitochondria in the cytoplasm of the cell. Different species of animals and plants have different numbers of chromosome pairs; for example, a mouse has 20 pairs and a tomato 12 pairs. As mentioned above, humans have 23 pairs, of which two, × and Y, determine gender. The chromosomes are homologous: there are two of each type (autosomes) except the X and Y (sex chromosomes). Each nucleated cell of the organism has the same DNA as every other cell unless a mutation or chromosomal rearrangement occurred after conception.
The chemical bonds of DNA bind each linear strand to the linear strand of the same sequence positioned in the reverse direction. Hydrogen bonding between adenine (A, a purine) from one strand and thymine (T, a pyrimidine) from the other, or guanine (G, a purine) and cytosine (C, a pyrimidine) maintains the alignment of the two strands. The tertiary structure of the double strand is a double helix. Since the pairing of A with T and G with C is essential to the secondary and tertiary structures, the two strands are complementary.
During the process of cell division (mitosis), all DNA is duplicated and each daughter cell receives the same information from the parent unless a mutation or chromosomal rearrangement occurs. The process of gamete (spermatozoa or ova) formation (meiosis) involves a halving of the number of chromosomes. During meiosis, a single cell forms four gametes, each of which has half the number of chromosomes (haploid). Crossing over (exchange of DNA or recombination) occurs during the duplication process between homologous chromosomes (a pair with the same gene loci in the same order), and genetic material is exchanged (Fig. 1.3). This process changes the order of the alleles on a chromosome and increases genetic variability. During fertilization, the two haploid cells (egg and sperm) fuse to form a diploid cell, restoring the normal number of chromosomes.
Single-gene defects may be caused by a point mutation that causes an alteration of an amino acid in the sequence of the protein product or by a deletion or duplication of DNA within the locus of a single gene. Point mutations of functional significance to the organism usually occur in an exon or regulatory sequence. Chromosomal abnormalities involve deletions (loss of material) or duplications (extra material) of DNA and can sometimes be identified microscopically.
Such abnormalities involve a larger segment of DNA or an entire chromosome. Haploinsufficiency is a gene-dosage effect that occurs when a diploid requires both functional copies of a gene for a wild-type phenotype. An organism that is heterozygous for a haploinsufficient locus does not have a wild-type phenotype.
Such abnormalities involve a larger segment of DNA or an entire chromosome. Haploinsufficiency is a gene-dosage effect that occurs when a diploid requires both functional copies of a gene for a wild-type phenotype. An organism that is heterozygous for a haploinsufficient locus does not have a wild-type phenotype.
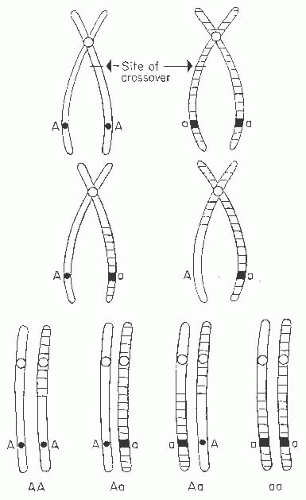 FIGURE 1.3. Crossing over (exchange of genetic material) between homologous chromosomes during meiosis. |
Tjio and Levan (10) identified the correct number of human chromosomes as 46; previously the total was thought to be 48. The normal human 22 pairs of autosomal chromosomes and one pair of gender-determining chromosomes (XY for male and XX for female) are divided into seven groups on the basis of length and centromere position. In 1960, the initial Denver classification was developed at a meeting in Colorado and was based on the overall length and centromere position; seven groups, labeled A to G, were created. In 1971, the Paris nomenclature was created, and each chromosome was identified by length, centromere position, and banding. Chromosomes 1,2, and 3 constituted group A; 4 and 5, group B; 6 to 12 and X, group C; 13 to 15, group D; 16 to 18, group E; 19 and 20, group F; and 21, 22, and Y, group G (Fig. 1.4).
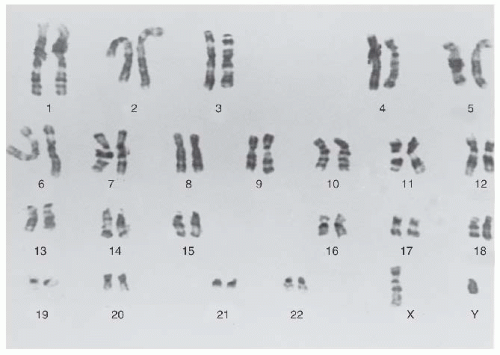 FIGURE 1.4. Karyotype of a normal male, showing banding produced by trypsin treatment of Seabright (11). (Courtesy of Robert S. Sparkes, MD.) |
Changes of chromosome structure can involve single chromosomes or an exchange of material between chromosomes. A piece of a chromosome may be lost by deletion or may be duplicated. The former results in monosomy for a group of genes and the latter results in trisomy for the genes. Chromosome segments can also be inverted—flipped 180 degrees from their normal orientation. If no material is gained or lost, the changes may not have a phenotypic effect. There are vast amounts of genetically inert DNA between groups of genes so usually these breaks cause no change in phenotype. Rarely, a gene may be disrupted by the chromosome breakage involved in the inversion. Another intrachromosomal rearrangement is the formation of a ring. This usually arises from breakage of the two ends and their subsequent fusion into a ring structure. There may be phenotypic consequences from deletion of chromatin from the two ends, and also from mitotic instability of the rings, resulting in trisomic or monosomic cells.
Translocation involves the exchange of material between chromosomes. Usually, translocations arise as reciprocal exchanges. If no material is lost or gained, the translocation is said to be balanced. Balanced translocations—and inversions for that matter—are occasionally found as variants in
the general population. It is estimated that approximately 0.2% of individuals carry an asymptomatic rearrangement. If one comes to medical attention, it is usually because the unbalanced offspring of an individual with a translocation has congenital anomalies, or the individual has a history of spontaneous abortions.
the general population. It is estimated that approximately 0.2% of individuals carry an asymptomatic rearrangement. If one comes to medical attention, it is usually because the unbalanced offspring of an individual with a translocation has congenital anomalies, or the individual has a history of spontaneous abortions.
CYTOGENETIC TESTS
Karyotyping
By synchronizing the reproductive cycle of a group of cells and arresting the progression of the mitotic process, the chromosomes can be visualized microscopically, and specific chromosomal identification can be made on the basis of length and the use of stains such as trypsin-Giemsa (G-banding; Fig. 1.4) (11), quinacrine mustard (Q-banding) (12), “reverse” or R-banding (Giemsa staining following controlled denaturation by heat), silver (staining of nucleolar organizing regions), and C-banding (staining of the condensed chromosome material near the centromere and regions with heterochromatin). All methods identify bands or specific regions and are useful for studying the specific structure of chromosome(s). The number of chromosomes can be counted and the bands of each studied for deletions, duplications, and other rearrangements. This technique is called karyotyping.
Relatively new techniques for arresting the progression of mitosis earlier in the cell cycle have been developed. In late prophase or early metaphase, the chromosomes are longer and less condensed and bands are further subdivided; smaller deletions and/or duplications can be detected. Extended chromosome analysis, termed high-resolution banding, is much more time-consuming but is particularly useful if a specific chromosomal abnormality or rearrangement is suspected. As the method is more labor intensive and expensive, one would not order high-resolution banding for a routine sample.
A cytogenetic nomenclature system has been adopted to describe the human chromosomal complement and indicate deviations from normal. An extra chromosome is indicated by a plus (+) and a missing one by a minus (-); thus 47,XX, +21 is a female with trisomy 21 (three copies of chromosome 21). The short arm of a chromosome is called p and the long arm is q. Chromosomal bands are numbered according to landmarks starting from the centromere up the short arm and down the long arm. Chromosomal rearrangements are described by noting the rearrangement and indicating the breakpoint(s). For example, a female with a deletion of the short arm of chromosome 4 with breakpoint at band p15 has the karyotype: 46,XX,del (4)(p15). A ring is indicated as r (e.g., 46,XY,r[13]male with a ring chromosome 13) and translocation as t (e.g., 46,XX,t[3;9][p14; q21] female with a translocation between chromosomes 3 and 9 with breakpoints at band p14 on chromosome 3 and band q21 on chromosome 9). An inversion is indicated as inv (e.g., 46XY,inv[2][p12q12]male with an inversion of chromosome 2 with breakpoints at p12 and q12).
Fluorescent In Situ Hybridization
Recently, DNA probes have become available for all human chromosomes—greatly expanding the field of molecular cytogenetics. If the origin of a duplication or translocation is unknown, the DNA probes can be fluorescently labeled and “painted” onto the metaphase spread or interphase nuclei chromosomal preparation, a technique called fluorescent in situ hybridization (FISH); chromosomal abnormalities can be readily identified under the microscope.
Telomeres, the physical ends of linear eukaryotic chromosomes, are specialized nucleoprotein complexes that have important functions, primarily in the protection, replication, and stabilization of the chromosome ends. In most organisms studied, telomeres contain lengthy stretches of tandemly repeated simple DNA sequences composed of GC-rich strands (called terminal repeats). These terminal repeats are highly conserved; in fact, all vertebrates appear to have the same simple sequence repeat in telomeres: (TTAGGG)n. Often sequences adjacent to the telomeric repeats are highly polymorphic, are rich in DNA repetitive elements (termed subtelomeric repeats), and in some cases, genes have been found in the proterminal regions of chromosomes.
Telomerase is the reverse transcriptase enzyme responsible for the extension of telomeric repeat sequences in most species studied. If telomerase activity is diminished or absent, telomeres will shorten. Shortened telomeres appear to lead to cell senescence (13). Eventually, telomeric sequences can shorten to the point where they are not long enough to support the telomere-protein complex protecting the ends and the chromosomes become unstable. These shortened ends become “sticky” and promote chromosomal rearrangements (14,15). Some rearrangements may contribute to the development of cancers (16,17). Telomere testing has been shown to identify alterations in 7% to 10% of cases with moderate/severe mental retardation and cases of multiple congenital anomalies with mental retardation in the setting of normal karyotype testing (18). The analysis involves the detection of deletions, duplications, or cryptic translocations using subtelomere FISH probes on each chromosome (15).
Comparative Genomic Hybridization
Comparative genomic hybridization (CGH) was developed to screen the entire genome for DNA content differences by comparing a test sample to a control (19,20). Because metaphase chromosomes are used as the substrate for analysis, the detection of unbalanced alterations is limited to the resolution of the metaphase target (at the level of a 450-band karyotype, approximately 5- to 10-Mb change). DNA microarray CGH is a powerful new technology capable of identifying chromosomal imbalance at a high resolution by cohybridizing differentially labeled test and control DNA samples to a microarray chip (21,22,23). The chip (small metallic platform with applied spots of known large-insert
DNA clones such as bacterial artificial chromosomes) technology offers higher resolution, is faster, and is highly sensitive. Because the DNA clones have a known map location and information, detected alterations are immediately linked to known genetic markers (pieces of DNA with a known chromosomal location and sequence), and the genetic location of a chromosomal abnormality such as a deletion or duplication can be determined by the map distances between known markers or by the length of the clones used.
DNA clones such as bacterial artificial chromosomes) technology offers higher resolution, is faster, and is highly sensitive. Because the DNA clones have a known map location and information, detected alterations are immediately linked to known genetic markers (pieces of DNA with a known chromosomal location and sequence), and the genetic location of a chromosomal abnormality such as a deletion or duplication can be determined by the map distances between known markers or by the length of the clones used.
Whole-Exome and Whole-Genome Sequencing
Newer techniques of producing multiple copies of the coding regions of the genome (exomes), or the entire genome including exons and intronic sequence (whole genome) have revolutionized gene variant detection in disease (www.ambrygen.com/exome-sequencing). There are many factors that make exome sequencing superior to single-gene analysis including the ability to identify mutations in genes that were not tested due to an atypical clinical presentation, or the ability to identify clinical cases where mutations from different genes contribute to the different phenotypes in the same patient. This technology is particularly useful for gene discovery in those conditions in which mapping has been confounded by locus heterogeneity and uncertainty about the boundaries of diagnostic classification. Direct-to-consumer whole exome sequencing services are available to individuals without a physician intermediary through programs such as 23andMe (www.23andme.com/).
SINGLE-GENE MUTATIONS
The substitution, addition, or deletion of one or more of the bases in an exon alters the DNA sequence and results in the formation of a protein with an abnormal amino acid at a specific site or the absence of the protein. Such abnormalities are believed to be common, and each human being is estimated to have three to five such mutations (24). If such a point mutation occurs in a portion of the protein that is not critical to function, the abnormality goes undetected (because it is not evident phenotypically) and does not affect the individual. If the codon error results in an amino acid substitution that changes the formation or function of the protein or truncates the product, the mutation is evidenced by reduced or (rarely) improved function, which would be advantageous to the organism from an evolutionary standpoint.
Autosomal Recessive Inheritance
For some mutations, the loss of a functional protein product from one of the chromosomes is asymptomatic because the gene on the homologous chromosome produces a normal product. If 50% activity of a protein is sufficient for normal function, the individual with one mutation is called a carrier, the signs and symptoms of the disease would be evident only in those who have two abnormal genes. Such a disorder is called autosomal recessive and is evident in some (statistically 25%) of the offsprings of two carriers (Fig. 1.5). In general, there is a higher incidence of parental consanguinity associated with rare autosomal recessive disorders. For some diseases, carriers probably have a common ancestor because nearly all affected individuals have the identical mutation; an example of this founder effect is Tay-Sachs disease (OMIM 272800: infantile developmental delay, paralysis, dementia, blindness with lipid-laden ganglion cells leaving a central “cherry-red” spot funduscopically, premature death before the age of 5 years). Mutations of the implicated gene, the enzyme hexosaminidase, occur more commonly in the Ashkenazi Jewish population.
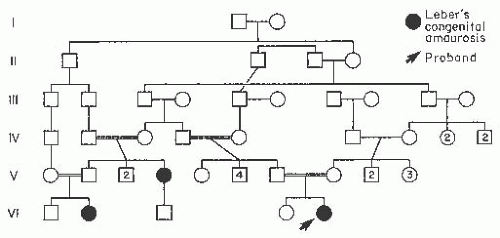 FIGURE 1.5. Pedigree of a family in which Leber congenital amaurosis (LCA) was inherited as an autosomal recessive disorder. Consanguineous matings are indicated by double lines. |
Autosomal Dominant Inheritance
When the protein is structural or the organism requires 100% activity for normal structure or function, phenotypic abnormalities are evident with a mutation of only one of the two homologous chromosomes. Such a disorder is termed autosomal dominant because the presence of a mutation on one of the chromosomes results in a phenotypically identifiable disease state. An affected individual with an autosomal dominant trait has a 50% chance of passing the gene to each offspring (Fig. 1.6). Examples of autosomal dominant disorders include Marfan syndrome (OMIM 154700: dislocated lenses, dilated aortic root, increased height, disproportionately long limbs and digits, anterior chest deformity, joint laxity, narrow arched palate, scoliosis due to connective-tissue disorder; caused by mutations of the fibrillin-1 gene); neurofibromatosis type I (OMIM 162200: consistent features of cafe au lait spots, Sakurai-Lisch nodules of the iris, fibromatous skin tumors, occasional hamartomatous tumors found systemically; due to mutations in the neuronbromin gene); and Best disease (OMIM 153700: mutations in the bestrophin gene cause
juvenile-onset and adult vitelliform macular dystrophy with collections of lipofuscin-like material in the subretinal space creating macular lesions which resemble an egg yolk). Disease-causing genes located on the autosomes occur equally in males and females, regardless of dominant or recessive inheritance.
juvenile-onset and adult vitelliform macular dystrophy with collections of lipofuscin-like material in the subretinal space creating macular lesions which resemble an egg yolk). Disease-causing genes located on the autosomes occur equally in males and females, regardless of dominant or recessive inheritance.
 FIGURE 1.6. A: Pedigree illustrating the autosomal dominant transmission of Waardenburg syndrome. (Adapted from DiGeorge AM, Olmstead RW, Harley RD. Waardenburg’s syndrome. A syndrome of heterochromia of the irides, lateral displacement of the medial canthi and lacrimal puncta, congenital deafness, and other characteristic associated defects. J Pediatr 1960;57:649-669.) (Figure 1.6 B) |
X-Linked Inheritance
Mutations on the X chromosome are unique, as normal females have a pair, and normal males have only one X chromosome along with an unpaired Y chromosome. X-linked diseases may be recessive or, much less frequently, dominant. Therefore, one abnormal gene for which an individual requires only some of the protein product usually would not cause disease in a female, who is a heterozygote, but would in a male, who is a hemizygote. Such disorders are termed X-linked recessive and become clinically evident in the male as he has only one X chromosome copy. Occasionally, a heterozygous female carrier exhibits some manifestations of an X-linked recessive disease. The expression of the single recessive gene on the × chromosome in a female has been explained by the X inactivation theory, or Lyon hypothesis (25). During the second week of embryonic life, one of the two X chromosomes in each cell of the female fetus randomly becomes the “inactive X”; this X chromosome becomes condensed during interphase and appears as a darkly stained mass at the nuclear membrane (Barr body or sex chromatin; Fig. 1.7). Most of the genes on the inactive X are not expressed. Once this differentiation occurs, the same X chromosome continues to be the inactive one in all the linear descendants of that cell. Thus, the female is a mosaic of two cell lines, those in which the genes on the maternally inherited X are active and those in which the paternally inherited X are active. The Lyon hypothesis is invoked to explain the splotchy pigment epithelium and choroidal pigmentation in the fundus of a female carrier of ocular albinism (OA), or the tapetal reflex in the carrier of X-linked retinitis pigmentosa (ghr.nlm.nih.gov/glossary=xch romsomeinactivation).
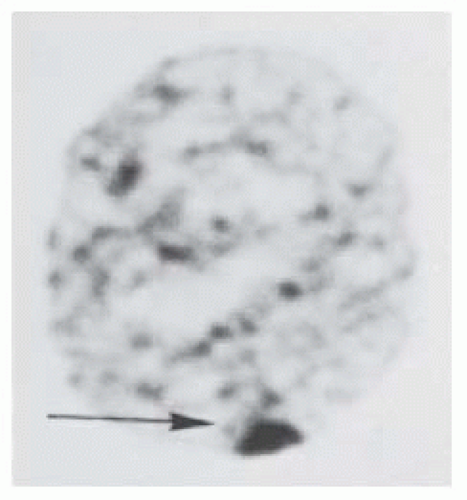 FIGURE 1.7. Barr body (sex chromatin) at the nuclear membrane in a cell from buccal mucosa of a normal female. |
The heritable inability to correctly perceive the color green, known as Daltonism (after the English chemist John Dalton, who himself was affected), was the first human trait mapped to the X chromosome (www.colblindor.com/2006/04/09/daltonism). X-linked disorders of the eye for which phenotypic evidence of the carrier state may be present in females include choroideremia (OMIM 303100: degeneration of the choriocapillaris and retina due to mutations in the Rab escort protein-1 gene [REP1]); Nance-Horan or cataract-dental syndrome (OMIM 302350: affected males have dense nuclear cataracts and frequent microcornea, and carrier females show posterior Y-sutural cataracts with small corneas and only slightly reduced vision; caused by mutations in the NHS gene) (26,27); blue cone monochromatism (OMIM 303700: affected males have poor central vision and color discrimination, infantile nystagmus, and nearly normal retinal appearance due to mutations in the locus control region upstream to the red and green cone pigment gene array) (28); and Lowe syndrome (OMIM 309000: affected males have cataracts, developmental delay, vitamin D-resistant rickets, and aminoaciduria, and carrier females have peripheral cortical lens opacities) (29). A carrier female has a 50% chance of passing a mutant gene on the X chromosome to any offspring; thus, the daughters have that same chance of being carriers and the sons of being affected with the disease (Fig. 1.8). Examples of X-linked recessive disorders include Duchenne muscular dystrophy (OMIM 310200: due to mutations of the dystrophin gene, affected males display progressive proximal muscular dystrophy with characteristic pseudohypertrophy of the calves and severe cardiomyopathy; there is massive elevation of creatine kinase levels in the blood, myopathic changes by electromyography, and myofiber degeneration with fibrosis and fatty infiltration on muscle biopsy); and X-linked juvenile retinoschisis (OMIM 312700: affected males with RS gene mutations have intraretinal splitting due to degeneration). If the X-linked gene is dominant, both the heterozygous female and hemizygous male manifest
the mutant phenotype. It has been observed in the few X-linked dominant traits that a heterozygous female is more likely to have female offspring, and it has been postulated that there is fetal wastage of males. Presumably, the hemizygous state of the mutation may be lethal. Examples of X-linked dominant disorders include familial incontinentia pigmenti type II (OMIM 308300: mutations in the IKK-gamma gene causing skin defects of perinatal inflammatory vesicles, verrucous patches, dermal scarring, and retinal vascular anomalies) and Aicardi syndrome (OMIM 304050: affected females have flexion spasms and lacunar lesions of the choroid and retina).
the mutant phenotype. It has been observed in the few X-linked dominant traits that a heterozygous female is more likely to have female offspring, and it has been postulated that there is fetal wastage of males. Presumably, the hemizygous state of the mutation may be lethal. Examples of X-linked dominant disorders include familial incontinentia pigmenti type II (OMIM 308300: mutations in the IKK-gamma gene causing skin defects of perinatal inflammatory vesicles, verrucous patches, dermal scarring, and retinal vascular anomalies) and Aicardi syndrome (OMIM 304050: affected females have flexion spasms and lacunar lesions of the choroid and retina).
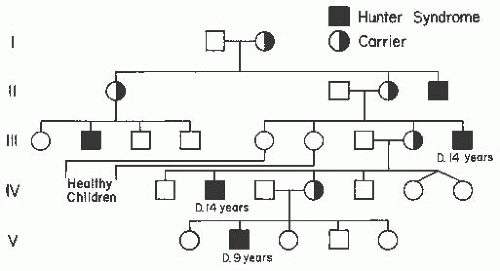 FIGURE 1.8. Pedigree showing X-linked inheritance of Hunter syndrome. Note that affected males are related through their mothers. |
Mitochondrial Genetics
An additional source of genetic information in the cell lies in the mitochondria (30). Each cell contains hundreds of mitochondria, each of which contains multiple copies of a 16,569base pair circular, double-stranded DNA molecule. This DNA encodes 13 peptides that are subunits of proteins required for oxidative phosphorylation. In addition, there is a complete set of 22 tRNAs and two rRNAs. These RNAs are involved in translation of mitochondrial-encoded proteins within the mitochondrion. Mitochondria are responsible for the generation of ATP via aerobic metabolism. Most mitochondrial proteins are encoded by nuclear genes; however, some are encoded by mitochondrial genes, and mutations can lead to energy failure. The mitochondrial genome is subject to a number of peculiarities of inheritance including maternal transmission and a phenomenon known as heteroplasmy, resulting in distinctive patterns of familial disease. Heteroplasmy determines expression variability of the disease within a family. Different cells in an individual and different individuals in a family contain different proportions of mutant and wild-type mitochondria (31).
Mitochondrial mutation disorders display maternal genetic transmission. There may be the appearance of transmission directly from generation to generation, suggesting dominant inheritance. Both males and females may be affected, but men never transmit the disorder to any of their offspring. Women, on the other hand, pass the trait to all of their children, although expression may be more severe in some than in others. At the time of fertilization, the sperm sheds its tail, including all of its mitochondria. Only the sperm head, containing nuclear DNA, enters the egg. Therefore, all the mitochondria for the next generation are contributed by the egg cell. Hence, mitochondrial genes are exclusively maternally derived, explaining the pattern of maternal transmission.
Major mitochondrial gene mutation syndromes include Kearns-Sayre (OMIM 530000: external ophthalmoplegia, pigmentary retinopathy, heart block, ataxia, increased cerebrospinal fluid protein); myoclonic epilepsy with ragged red fibers or MERRF (OMIM 545000: myoclonic epilepsy, myopathy, dementia); mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes or MELAS (OMIM 540000: lactic acidosis, stroke-like episodes, myopathy, seizures, dementia); Leber hereditary optic neuropathy (OMIM 535000: blindness, cardiac conduction defects); and Leigh syndrome (OMIM 256000: movement disorder, respiratory dyskinesia, regression) (32,33,34,35,36).
Penetrance
The clinical features of single-gene mutations vary from one individual to another. Penetrance is the percentage of known carriers manifesting the phenotype and reflects our ability to identify an individual with a mutant gene; reduced penetrance means that some individuals with the gene may not exhibit clinical evidence of the disease. Nonpenetrance is defined as the absence of phenotype in a person known to carry a specific mutant gene. Nonpenetrance has been demonstrated to occur with many genetic traits and can be most easily inferred when a grandparent and child have a disorder that does not appear to be expressed in the parent. For example, some individuals without evidence of colobomatous microphthalmia have an affected ancestor and an affected child (Fig. 1.9). The only potential explanation is that the apparently unaffected individual has the mutant gene in his or her genome but that the clinicians cannot find the evidence by ocular examination. If four family members descended from the same affected ancestor had affected progeny but one was free of the disease, the gene would be 75% penetrant.
Expressivity
Expressivity refers to the degree of phenotypic expression of a genetic trait. A disease may exhibit different manifestations among individuals in the same family. Such variable expressivity is common, particularly in autosomal dominant disorders. For example, some individuals with Marfan syndrome (OMIM 154700), an autosomal dominant disease of the connective-tissue gene fibrillin, may be tall and have dislocated lenses; others may be of normal stature and show no evidence of a dislocated lens but have an aneurysm of the ascending aorta.
Autosomal recessive disorders can exhibit variable expressivity, particularly if the mutation occurs at different sites in the protein. For example, there are numerous hemoglobinopathies with distinctive clinical manifestations that are caused by different mutations of the hemoglobin gene.
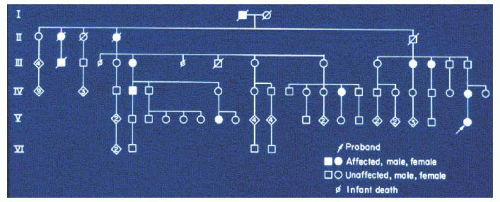 FIGURE 1.9. Multigenerational family with autosomal dominant colobomatous microphthalmia. The proband’s (arrow) maternal grandfather was examined and showed no evidence of colobomata or microphthalmia. Thus, in this family the gene for colobomatous microphthalmia exhibits reduced penetrance. |
Genetic Heterogeneity
Different gene defects, modes of inheritance, and chromosomal abnormalities can produce similar clinical phenotypes, a phenomenon termed genetic heterogeneity (ghr.nlm. nih.gov/glossary=geneticheterogeneity). Examples of genetic heterogeneity are common in ophthalmology. Classification of diseases is thus most reliably made on the basis of specific gene mutations or chromosomal aberrations; doing so allows for directed therapies.
Epigenetics
Epigenetics is the study of changes in gene expression or cellular phenotype caused by mechanisms other than changes in the original DNA sequence (www.pbs.org/wgbh/nova/body/epigenetics.html, and learn.genetics.utah.edu/content/epi genetics/). The development and maintenance of an organism is orchestrated by a set of chemical reactions that switch parts of the genome off and on at strategic times and locations. Epigenetics is the study of these reactions and the factors that influence them. In epigenetic modification, a methyl group (-CH3) is added to specific cytosine bases of the DNA. This enzymatic process, called DNA methylation, is known to play a key role in both development and disease. Methylation is a physical modification to the DNA that affects the way the molecule is shaped and, consequently, regulates which genes are transcriptionally active. Another epigenetic modification of DNA is the addition of a hydroxymethyl group (-CH2-OH) to specific cytosine bases of DNA. In eukaryotic cells, genomic DNA is wrapped around histone proteins to form nucleosomes, which together form chromatin fibers that can be further compacted into dense coils. Epigenetic modifications to histone proteins can either inhibit or promote coiling or condensation of the chromatin, leading to chromatin organizational changes, which in turn affects how the associated genes are expressed.
Mucopolysaccharidosis
The autosomal recessive Hurler syndrome or mucopolysaccharidosis type IH (OMIM 607014: corneal clouding, coarse facies, developmental delay, hepatosplenomegaly, and hernia) and X-linked recessive Hunter syndrome or mucopolysaccharidosis type II (OMIM 309900: no corneal clouding, coarse facies, dwarfism, hepatosplenomegaly, and deafness) may be difficult to differentiate clinically on the basis of physical findings in a young boy with no family history. As the inheritance patterns are different, the gene defects are distinctive (http://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i). Conversely, the clinical features of the Hurler and Scheie (OMIM 607016: stiff joints, clouding of the peripheral cornea, and aortic regurgitation) syndromes can be differentiated by the mental retardation and early death that are features of the Hurler syndrome. Life expectancy in the Scheie form is nearly normal and mental retardation is very rare. Originally the two were classified as different diseases. With the identification of the enzymatic defect, it became clear that the disorders were allelic as the same gene is mutated in both, causing a deficiency in the enzyme alpha-1-iduronidase.
Leber Congenital Amaurosis
Leber congenital amaurosis (LCA; OMIM 204000) is a group of autosomal recessive retinal dystrophies. It is the most common genetic cause of congenital retinal disorders in infants and children, and results in significant and often severe vision loss at an early age. Its incidence is 2 to 3 per 100,000 births and it accounts for 10% to 18% of cases of congenital blindness. At least 17 genes contribute to this disorder (37). The observed genetic heterogeneity is the result not only of the number of genes that have been implicated in LCA, but also the consequences of the different mutations in these genes. Mutations in at least two of these genes—RPE65 and CRX—not only cause an LCA clinical presentation, but also lead to other late-onset retinal dystrophies such as retinitis pigmentosa and cone-rod dystrophy (38,39). Gene therapy was used to successfully recover vision in a canine model of LCA (40). The researchers designed an adeno-associated virus vector to use in retinal transgene delivery. This vector was used to
carry wild-type canine RPE65 complementary DNA and was injected into the subretinal space of dog eyes. The LCA dogs showed visual recovery in performing visual function and behavioral tests. Multiple gene therapy trials are in progress to treat humans with the same genetic disease (37).
carry wild-type canine RPE65 complementary DNA and was injected into the subretinal space of dog eyes. The LCA dogs showed visual recovery in performing visual function and behavioral tests. Multiple gene therapy trials are in progress to treat humans with the same genetic disease (37).
Albinism
Albinism comprises a group of heterogeneous inherited abnormalities of melanin synthesis characterized by a congenital reduction or absence of melanin pigment associated with specific developmental changes of the visual system (www. albinism.org). Oculocutaneous albinism (OCA) involves two regions of the body: the skin and hair and the optic system, including the eye and optic nerves. Ocular albinism (OA) has similar changes in the visual system by reducing mainly the pigment in the retinal pigment epithelium (RPE) of the eye, usually with no clinical difference in the color of the skin and hair. Ophthalmologic signs, although variable, include nystagmus, hypopigmentation of the uveal tract and RPE, iris transillumination, foveal hypoplasia, and abnormal decussation of the optic nerve fibers at the optic chiasm.
The formation of melanin pigment is a complex event requiring several enzymes and proteins and the pigmentcontaining subcellular organelle, the melanosome. Melanin pigment is produced in the melanocyte, which is found in the skin, hair follicles, iris, and RPE of the eye. Melanin biosynthesis begins with hydroxylation of the amino acid 1-tyrosine to dihydroxylphenylalanine (DOPA) and the oxidation of DOPA to DOPA quinone by the copper-containing enzyme tyrosinase, resulting in either black-brown eumelanin, or in the presence of sulfhydryl compounds, red-yellow pheomelanin. The resulting pigment polymer is deposited on a protein matrix within the melanosome. In the skin and hair follicles, the melanosome is then transferred to keratinocytes via the dendrites of the melanocyte.
At present, four genetic loci responsible for human albinism have been mapped (OCA1: OMIM 203100, OCA2: OMIM 203200, Hermansky-Pudlak syndrome: OMIM 203300, and OA1: OMIM 300500); three of the genes have been isolated and pathologic mutations identified (OCA1, OCA2, and OA1). The classic “tyrosinase-negative” albinism results from tyrosinase gene mutations that inactivate the encoded enzyme and is categorized as OCA1 (41). Mild forms of OCA1, initially described as autosomal recessive OA, result from mutations of the tyrosinase gene that produce an enzyme with residual activity. OCA in which hairbulbs form melanin upon incubation in DOPA or tyrosine (tyrosinase-positive OCA) can be caused by mutations at several loci, including OCA1; OCA2, which is associated with the human homolog (P) of the mouse p gene— a melanosomal transport protein (42); and the rare Hermansky-Pudlak syndrome (OCA with platelet dysfunction).
Magnetic resonance imaging (MRI) size and configuration comparisons of the optic pathways in normal versus albinism phenotypes revealed significantly smaller optic nerves and tracts, smaller chiasmatic widths, and wider angles between nerves and tracts in the albino group than in the control group (43). The chiasms of the albinos are shaped like an X, whereas the chiasm in the controls was shaped like two back-to-back parentheses, that is, “)(.” These differences reflect the atypical crossing of optic fibers. MRI can be an important diagnostic tool in patients with equivocal albinotic presentations.
X-linked OA1 is caused by mutations in the OA1 gene at chromosome Xp22.3-p22.2, which encodes a membrane glycoprotein localized to melanosomes. Approximately 48% of the reported mutations in the OA1 gene are intragenic deletions and about 43% are point mutations. Faugere and associates (44) report three OA1 unrelated families with an initial diagnosis of congenital nystagmus. They identified three novel OA1 mutations consisting of two intragenic deletions and a point mutation. Direct testing of carrier females is advocated and can be performed by direct sequencing or by scanning methods such as denaturing gradient gel electrophoresis or denaturing high-performance liquid chromatography. The real-time fluorescent PCR gene-dosage assay is an accurate, nonradioactive, and fast method for gene carrier assessment that can be applied to any type of OA1 gene deletion. A similar two-tiered diagnostic test strategy for mutation screening for OA1 has been proposed (45).
Examples of Pleiotropic Disorders with Ophthalmic Considerations
A single-gene mutation that is expressed in many different tissues or can affect more than one organ system is termed pleiotropic. Waardenburg syndrome (Ptosis-epicanthus syndrome) is an example of a disorder in which the mutation of one gene has multiple organ effects. This dominantly inherited disorder includes displaced canthi, heterochromia of the irides, white forelock, broad nasal root, and deafness (Fig. 1.6); the syndrome is divided into four types (I, II [A, B, and C], III, and IV) with the most severe being associated with upper limb defects (type III; OMIM 148820) and a ganglionic megacolon (Hirschsprung disease) (type IV; OMIM 277580). Dystopia canthorum, the lateral displacement of the inner canthi, distinguishes type I (OMIM 193500) (which shows this clinical phenotype) from type II. Types I and III (OMIM 148820) are caused by mutations in the PAX3 gene, which was first identified in Drosophila (46,47). Mutations in the transcription factor MITF have been implicated for Waardenburg syndrome type IIA (OMIM 193510) (48,49,50); digenic inheritance of type IIA and autosomal recessive OA has been proposed (51). Type IV or Waardenburg-Shah syndrome is a disorder of the embryonic neural crest that combines clinical features of Waardenburg syndrome and Hirschsprung disease with colonic aganglionosis.
Alagille Syndrome
Alagille syndrome (OMIM 118450) is an autosomal dominant disorder characterized by cholestatic liver disease, pulmonic valvular and peripheral arterial stenosis, “butterfly” vertebrae, posterior embryotoxon (anterior displacement of Schwalbe’s
line) with retinal pigmentary changes in the eye in some individuals, and unusual facies with broad forehead, pointed mandible, and bulbous nose tip (http://digestive.niddk.nih.gov/ddiseases/pubs/alagille/). The syndrome is due to mutations in the Notch signaling JAG1 gene on chromosome 20p12 (52,53).
line) with retinal pigmentary changes in the eye in some individuals, and unusual facies with broad forehead, pointed mandible, and bulbous nose tip (http://digestive.niddk.nih.gov/ddiseases/pubs/alagille/). The syndrome is due to mutations in the Notch signaling JAG1 gene on chromosome 20p12 (52,53).
Cornelia de Lange Syndrome
Cornelia de Lange syndrome (CDLS; OMIM 122470) is a dominantly inherited multisystem developmental disorder characterized by growth and cognitive retardation, abnormalities of the upper limbs, gastroesophageal dysfunction, hirsutism, cardiac and genitourinary anomalies, and characteristic facial features (54,55,56) (www.cdlsusa.org/). Ophthalmic features include ptosis, nasolacrimal duct obstruction, arched eyebrows with synophrys, long eyelashes, refractive error, and infrequent glaucoma (57). The prevalence is estimated to be as high as 1 in 10,000. Gene mutations in the NIPBL gene on chromosome 5p13.1 have recently been associated with this disorder (58,59). The jly homolog of NIPBL, Nipped-B, facilitates enhancer-promoter communication and regulates Notch signaling and other developmental pathways in D. melanogaster (60).
Marfan Syndrome
Marfan syndrome (OMIM 154700) is an autosomal dominant connective-tissue disorder with an estimated incidence of 1 in 5,000 with approximately 25% being sporadic cases (www.marfan.org/). Three systems are predominantly involved: the skeleton, heart, and eye. Common and major manifestations of the disease include subluxation of the crystalline lens; dilatation of the aortic root and aneurysm of the ascending aorta; and skeletal abnormalities such as pectus excavatum and kyphoscoliosis and an upper segment/lower segment ratio of two standard deviations below the mean for age. Other criteria such as myopia, mitral valve prolapse, arachnodactyly, joint laxity, tall stature, pes planus, pneumothorax, and dural ectasia are also contributory. The diagnostic criteria have been recently revised, requiring involvement of three systems with two major diagnostic manifestations (61). The gene defect has been identified: Fibrillin (FBN1) maps to chromosome 15q15-q21.1. It is a large gene of more than 230 kb and highly fragmented with 65 exons. The protein is a 350-kDa glycoprotein and is the principal structural component of connective-tissue microfibrils found ubiquitously in all extracellular matrices. Fibrillin structures serve as scaffolds for the deposition of elastin in elastic tissues (61).
To date, more than 500 mutations have been identified in the FBN1 gene in Marfan syndrome patients and related diseases (61). Presently, no definite genotype-phenotype correlations have been identified except for neonatal mutations. An association with a subset of mutations in exons 24 to 32 and neonatal MFS appears correlative, thus molecular diagnostic testing can be performed (62). With a few exceptions, almost all mutations found in fibrillinopathies other than classic Marfan syndrome have been unique to one affected individual or family, which has hampered the delineation of potential genotype-phenotype correlations (62).
Comeglio and associates (63) characterized the incidence and class of FBN1 mutations in the largest series reported to date, a group of 11 consecutive British patients affected predominantly by ectopia lentis (EL). The investigators identified six causative mutations in the fibrillin 1 gene (FBN1)—three mutations are novel and one was recurrent in two patients—thus establishing an FBN1 mutation incidence of 63% (7/11). These results are within the 23% to 86% range of mutation identification rate in Marfan syndrome and related patients in recent investigations. All mutations were within the first 15 exons of the fibrillin gene, while database citations of Marfan mutations are distributed throughout the gene. A different type of FBN1 mutation presents in this group of patients, with arginine to cysteine substitutions appearing frequently. Patients with predominant EL should be screened for FBN1 mutations. Echocardiography is recommended initially and at regular intervals throughout the patients’lifetimes, as there is a tendency for late-onset aortic dilatation and/or dissection to develop.
Lens subluxation is the diagnostic ocular abnormality in this disease. It is present in 65% to 70% of patients and varies from a mild superotemporal displacement of the lens seen only in postpupillary dilation to significant subluxation that places the equator of the lens in the pupillary axis. Subluxation of the lens due to stretched or absent zonular fibers is slowly progressive in the first two decades of life. Further displacement at a later age is uncommon. Total dislocation into the vitreous cavity is uncommon and may be complicated by phacolytic uveitis and glaucoma. Anterior displacement of the lens into the anterior chamber or within the pupillary space is rare in Marfan syndrome with EL. Premature cataracts are common, developing 10 to 20 years earlier in patients with Marfan syndrome than in the general population.
MASTER CONTROL GENES
The mature eye is a highly complex organ that develops through a highly organized process during embryogenesis. Alterations in the genetic programming of the eye could lead to outcomes of severe eye disorganization that are apparent at birth or shortly thereafter.
For the past decade, several master-control genes that direct developmental and differential pathways have been identified. PAX6, SOX2, and RX are at the top of the eye developmental hierarchy; mutation or loss of these genes leads mainly to loss of the entire eye. Other genes, such as FOX, PITX, and MAF, are important for the development of particular regions of the eye and are thought to be downstream of the top level of regulation in eye development. These genes are expressed during embryogenesis and initiate a cascade of gene expression responsible for specific cell-lineage commitments. Most genes at the top of the hierarchy of eye development code for transcription
factors, although a few code for signaling molecules. The mutant phenotypes that are associated with some of these genes are described below, along with the genetic and molecular interactions that have been inferred between their products.
factors, although a few code for signaling molecules. The mutant phenotypes that are associated with some of these genes are described below, along with the genetic and molecular interactions that have been inferred between their products.
Paired Box Gene 6
Paired box gene 6 (PAX6) is the prototype for an eye master-control gene. It is one of the family of genes that encode transcription factors with a homeodomain and a paired domain. Loss of function leads to the eyeless (ey) phenotype in Drosophila and also causes severe ocular defects in many other animals (64). Interestingly, ectopic (at a different location other than normal) expression of Pax6 in Drosophila, mouse, and frog (Xenopus) causes the formation of a functional eye. This supports two concepts: (a) homologous genes in different species have the same function—to “switch on” eye development; (b) there is only one way to make the eye, by a cascade of signals initiated by Pax6 expression (65).
In humans, PAX6 mutations mainly cause aniridia (OMIM 106210), which is a panocular disorder, and less commonly cause isolated cataracts and macular hypoplasia (failure of retinal foveal development; OMIM 136520), keratitis (OMIM 148190), and Peters’ anomaly (central corneal opacity which is frequently associated with adhesion between the cornea and the lens; OMIM 603807) (66). As in the mouse, the homozygous loss (both parental alleles have mutations) of PAX6 function in humans affecting all expressing tissues is lethal.
Targets of PAX6 encode other transcription factors or structural proteins of the lens (crystallins) and cornea (keratins 1-12). Many PAX6-regulated transcription factors (such as SIX3, FOXE3, MAF, MITF, PROX1, LHX2, and PITX3) are involved in the formation of the cornea and lens; others (such as PAX2, CHX10, and EYA1) are involved in retina and optic nerve development (67).
Only a few transcription factors or signaling molecules (e.g., BMP4, BMP7, RX, and SHH) are known to regulate PAX6. Among them, SHH might be highest in the hierarchy: knockout mice and humans with mutations in SHH suffer from holoprosencephaly with ocular manifestations that range from microphthalmia to cyclopia, which indicates a disruption in the earliest event in eye development, separation of the central eye field (68).
Genotype and phenotype information for human PAX6 mutations is freely available from the PAX6 mutation database (http://pax6.hgu.mrc.ac.uk).
Sex-Determining Region Y-Related High-Mobility Group Box Gene 2
Sex-determining region Y-related high-mobility group box gene 2 (SOX2) is expressed in the developing lens placode, lens pit, optic cup, neural retina, lens, brain, and ear; heterozygous mutations of the gene result in anophthalmia in humans. Interestingly, all of these mutations seem to occur de novo (spontaneously) and are inherited as dominant alleles (69).
Retina and Anterior Neural Fold Homeobox Gene
The retina and anterior neural fold homeobox gene (RX) encodes a homeodomain transcription factor and is one of the first retinal patterning genes to be expressed during development (70). Rx-deficient mouse embryos lack eye anlagen (simple preliminary organ structures in the embryo) and do not express Pax6 in the eye field, which indicates that Pax6 upregulation in these tissues is dependent on functional Rx expression (71). It has recently been shown that the spontaneous mouse mutant eyeless (analogous to anophthalmia in humans) is caused by a point mutation in an alternative translation-initiation codon of Rx (72).
ISOLATED OCULAR AND PERIOCULAR SYNDROMES
Microphthalmia and Anophthalmia
Microphthalmia (OMIM 309700), a term derived from the Greek micro (small) and ophthalmos (eye), refers to a congenital malformation in which the volume of the eye is reduced; the spectrum ranges from mild reduction in the anteroposterior axis to histologically documented anophthalmia (OMIM 206900) (www.ncbi.nlm.nih.gov/books/NBK1378/). Nanophthalmia (OMIM 600165 and 605738) is used to describe microphthalmia with normal intraocular structures, and refers to a refractive error of +8.00 D or greater hyperopia. Although microcornea or high hyperopia may be a useful clinical clue, the diagnosis frequently can be made by inspection alone; however, as microcornea can occur in the absence of microphthalmia (73,74) and conversely, microphthalmia in association with a normal-sized cornea (75,76,77), the clinical diagnosis may be inaccurate (http://eyepathologist.com/disease.asp?IDNUM=308040). As the spectrum of microphthalmia varies from slightly reduced axial length to histologically documented anophthalmia, ultrasonography with precise measurement of the anteroposterior axis is essential for the diagnosis. Since most normal adult eyes range from 21.50 to 27.00 mm, an adult axial length of less than 20 mm should be considered abnormal (78,79). Despite the use of this technologically advanced tool, extreme microphthalmia may be difficult to distinguish from anophthalmia.
Microphthalmia appears to be a relatively common ocular malformation in all races. The high prevalence, which would be unusual for a disorder caused by a single gene, suggests multiple causes. Few studies have documented the prevalence in the general population. In a prospective study of more than 50,000 pregnancies in the United States in the late 1960s, the incidence of anophthalmia or microphthalmia was found to be 0.22 per 1,000 births and that of coloboma to be 0.26 per 1,000 (80). Prevalence among blind adults varies from 0.6% to 1.9% (81,82). In the pediatric age group, it accounts for 3.2% to 11.2% of cases of blindness (82,83,84). These differences are not readily explainable
but may reflect the race or population studied; the highest prevalence (11.2%) was found in the 1980 survey on the causes of blindness among Japanese schoolchildren.
but may reflect the race or population studied; the highest prevalence (11.2%) was found in the 1980 survey on the causes of blindness among Japanese schoolchildren.
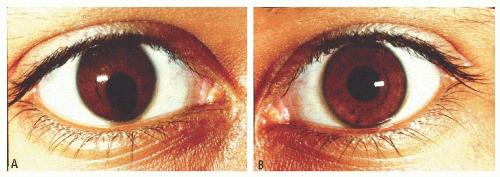 FIGURE 1.10. Right (A) and left (B) irides of a young man with mental retardation and deletion of the long arm of chromosome 18. Note the asymmetry. |
Visual impairment in microphthalmia varies from little or no loss to absence of vision, as would be found in cases of anophthalmia. The degree of visual loss best correlates with the associated abnormalities and degree of microphthalmia; cataracts, optic nerve hypoplasia, and/or colobomata of the macula or optic nerve may cause significant visual impairment.
Cases of microphthalmia may be divided into two general forms: colobomatous and noncolobomatous. Although uveal colobomata may occur in the absence of microphthalmia, the two are frequently associated and presumably etiologically related. A coloboma may occur in the iris, choroid, optic nerve, or any combination thereof (Figs. 1.10 and 1.11). The colobomata result from incomplete closure of the fetal fissure, a process normally completed by the 6th week of gestation (85). The embryonic processes that determine the size of the eye are poorly understood. Congenital cystic microphthalmia and anophthalmia are extreme forms of this dysembryogenesis (86,87). As extreme microphthalmia may be difficult to distinguish from anophthalmia, serial sectioning of the orbit may be necessary to differentiate them. The malformation may be unilateral or bilateral, and asymmetry is common.
Microphthalmia, with or without colobomata, may be a manifestation of many different disorders: genetic, environmental, and those of unknown cause. The evaluation of a patient is interdisciplinary. Aside from a complete ocular examination, all patients should have a careful history, including a genetic pedigree and physical examination. In certain cases, family members should also be examined.
Isolated colobomatous microphthalmia (OMIM 300345) may occur as an autosomal dominant disorder with incomplete penetrance; expressivity varies from small colobomata of the eye to microphthalmia and even anophthalmia (Fig. 1.9). Microphthalmia without colobomata also may be inherited as an autosomal dominant disease; more commonly, associated congenital cataracts or other ocular malformations are present (88,89). Microphthalmia with congenital retinal detachment or congenital cataracts may be inherited as an autosomal recessive disorder (90,91).
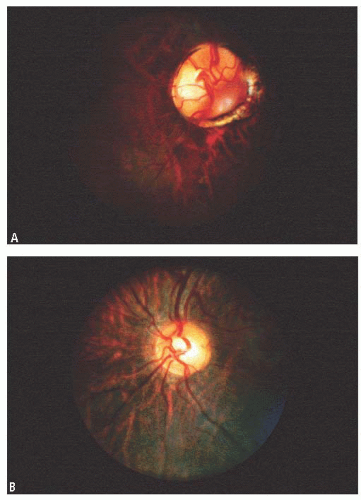 FIGURE 1.11. A: Coloboma of the right optic nerve in an eye with 20/20 vision. B: The left eye optic nerve head is normal. |
Microphthalmia (Mi) was one of the first mouse mutant genes to be described in which the development of the retina is affected. An interesting allelic series ranging from weak recessive to severe dominant phenotype has been compiled and genetically characterized. The eyes of the mutants develop poorly because of the defects in the RPE. The mutated gene, microphthalmia-associated transcription factor (Mitf) is a member of the basic-helix-loop-helix leucine zipper family of transcription factors. Mutations in the
human homolog MITF (OMIM 156845) cause 20% of cases of Waardenburg syndrome type IIA (OMIM 193510) (92).
human homolog MITF (OMIM 156845) cause 20% of cases of Waardenburg syndrome type IIA (OMIM 193510) (92).
Human mutations in SIX3 lead to holoprosencephaly; in some cases, the phenotype is milder and manifests as microphthalmia and iris coloboma (93). Several reports indicate that SIX3 might function as a repressor of some developmental processes in the eye (94,95). In the mouse, Six3 is activated by Pax6 and Prox1 (a transcription factor that is important for the differentiation of lens fiber cells and alpha crystalline expression) but is regulated by its own negative feedback loop (96). Transgenic mouse experiments have shown that Pax6 and Six3 regulate the transcription of each other (97).
The CHARGE syndrome (OMIM 214800) is characterized by the nonrandom association of colobomatous microphthalmia, heart defects, atresia choanae, retarded growth, genital anomalies, and ear anomalies or deafness; at least two of the features are necessary for the diagnosis (98,99,100,101,102,103,104) (www. chargesyndrome.org/). The ocular malformation is a common feature; the cardiac defects described in the syndrome are varied and may be lethal. The phenotypes of the chromosomal trisomy syndromes (13,18), 4p— (Wolf-Hirschhorn; deletion of the short arm of chromosome 4) syndrome, and cat-eye syndrome may be similar to the features of CHARGE association, and chromosomal analysis may be necessary to clarify the diagnosis (see section “Chromosomal Rearrangements”).
Two genetic loci have been identified in conjunction with isolated high hyperopia, autosomal dominant nanophthalmos (NN 01; OMIM 600165) on chromosome 11p (105) and NNO2 (OMIM 605738), which maps to chromosome 15q12-15. The phenotype for NNO2, however, is not of isolated, nonsyndromic high hyperopia. Rather it is that of unilateral or bilateral microphthalmos with variable expressivity such as corneal clouding and iridocorneal synechiae resembling Peters’ anomaly or no microphthalmia with optic nerve agenesis.
Ocular Adnexa and Eyelid Abnormalities
The development of the eyelids and ocular adnexal structures is closely related to the formation of the eyes themselves. Complete failure of the eyelids to form results in cryptophthalmos (OMIM 123570), a condition in which the skin extends from the forehead to the cheeks uninterrupted, but attached to a usually malformed globe underneath. It may occur in isolation or as part of Fraser syndrome (OMIM 219000), which is autosomal recessive. A review of 27 cases of isolated cryptophthalmos revealed 11 of those to be familial, inherited in a dominant fashion (106). A gene has not yet been identified. Treatment depends upon the functional capability of the underlying globe, and this can be measured using imaging modalities in conjunction with visually evoked response and electroretinogram testing. If visual potential can be demonstrated, surgery may be attempted with the goal of creating a clear visual axis, possibly via a keratoprosthesis, as well as creating functioning lid structures. There is a clinical trial for this and a closely related disorder, Fryns syndrome (clinicaltrials.gov/ct2/show/NCT00032877).
Eyelid formation during development requires the eyelid folds to initially fuse and then to separate into upper and lower lids. The failure of this separation results in ankyloblepharon (OMIM 106250 and 106260), a condition in which the eyelids are completely or partially joined. Filiforme adnatum is a unique condition characterized by multiple strands of tissue connecting the two eyelids.
Structural maldevelopment of the eyelid may result in an eyelid coloboma or a disruption in the margin of the eyelid. Isolated eyelid colobomata range from a near total absence of the lid to the appearance of a small notch in the lateral aspect of the lid. Colobomata of the eyelid may also be part of a syndrome such as Treacher Collins syndrome (OMIM 154500) or Goldenhar syndrome (OMIM 164210) or may occur in association with cleft palate, dermoid, cleft lip, microphthalmia, iris colobomata, brow colobomata, and osseous facial clefts.
Congenital ptosis is due to a deficiency of the striated muscle fibers in the levator muscle. This abnormality may occur in three main forms: simple hereditary ptosis, with external ophthalmoplegia, and in the blepharophimosis syndrome (horizontal narrowing of the palpebral fissure; OMIM 110100). Simple congenital ptosis, unilateral or bilateral, may be inherited in an autosomal dominant fashion with incomplete penetrance (107). A gene for hereditary congenital ptosis type 1 (OMIM 178300) has been mapped to 8q21.12 (108). A case of bilateral isolated ptosis was also reported in a male patient found to have a de novo balanced translocation t(1;8) (p34.3;q21.12). The cytogenetic breakpoints were refined, and the chromosome 8 breakpoint was found to disrupt a gene homologous to the murine zfh4 gene, which encodes for a transcription factor expressed in muscle and nerve tissue. This suggests the human ZFH4 may be a candidate gene for congenital bilateral isolated ptosis (109). Analysis of a large white English pedigree revealed an X-linked dominant form of congenital isolated ptosis that mapped to chromosome Xq24q27.1. This was named hereditary congenital ptosis type 2 (OMIM 300245) (110). Treatment for isolated congenital ptosis is surgical and may be performed on patients at any age; however, if the ptosis is significant, early surgery is necessary to prevent occlusion amblyopia. The choice of surgical procedure is dictated by the degree of ptosis, amount of levator muscle function, and clinical response to 2.5% phenylephrine ophthalmic solution. Patients who have a mild to moderate amount of ptosis which improves within 5 minutes after the administration of 2.5% phenylephrine ophthalmic drops into the upper eyelid fornix may benefit from a Mueller’s muscle resection, while those with minimal response may require a levator muscle resection. In the absence or near absence of levator muscle function, a frontalis sling procedure is performed.
The association of blepharophimosis with ptosis and epicanthus inversus (BPES) is usually inherited in an autosomal dominant pattern (111). There are two types of BPES— type I is associated with ovarian failure and type II is not. The gene, FOXL2, is a winged helix/forkhead transcription factor and maps to chromosome 3q22-q23 (112,113,114). FOXL2 is mutated to produce truncated proteins in type I
families and larger proteins in type II. In this disorder, the palpebral fissures are narrowed both horizontally and vertically, and the ptosis is characterized by poor levator muscle function and the absence of a lid crease. Treatment involves correcting the ptosis with a frontalis sling procedure due to the poor levator muscle function. Surgical repair of the epicanthus inversus may also be undertaken in some cases.
families and larger proteins in type II. In this disorder, the palpebral fissures are narrowed both horizontally and vertically, and the ptosis is characterized by poor levator muscle function and the absence of a lid crease. Treatment involves correcting the ptosis with a frontalis sling procedure due to the poor levator muscle function. Surgical repair of the epicanthus inversus may also be undertaken in some cases.
Fibrosis of the extraocular muscle syndrome (FEOM) is a congenital disorder of innervation to the extraocular and eyelid muscles, which in turn affects muscular development (115). Affected individuals have a nonprogressive inability to move some or all of the extraocular muscles due to fibrotic and scarred muscles, adhesions between muscles and Tenon’s capsule, and adhesions between Tenon’s capsule and the globe. The eyes are usually fixed in a downward gaze, and as a result the patient assumes a chin-up head posture. The adhesions may involve the levator muscle, resulting in bilateral ptosis. FEOM1 (OMIM 135700) is caused by a mutation in the KIF21A gene (116), typically maps to chromosome 12q12, and shows neuropathologic changes suggesting a primary defect in the development of the superior division of the oculomotor nerve (117,118). FEOM2 (OMIM 602078) is associated with bilateral ptosis, with the eyes fixed in an exotropic position. This autosomal recessive phenotype maps to chromosome 11q13.3—q13.4 and results from mutations of the ARIX gene (119). In families with FEOM3 (OMIM 600638), one or more affected individuals do not have the classic findings of the disorder. Their eyes may be not be infraducted or may elevate above midline, or the individual may be unilaterally affected. Ptosis may not be present (120).
Congenital entropion and ectropion refer to a malposition of the eyelid where the eyelashes are rotated towards and away from the globe, respectively. Congenital entropion results from imbalances in preseptal and pretarsal orbicularis. Congenital ectropion may result from inflammatory conditions such as Chlamydia infection and may be associated with noninflammatory conditions such as Down syndrome and ichthyosis.
Distichiasis, an anomaly in which two rows (instead of one row) of lashes are present at the lid margin, may be autosomal dominant. During development, eyelashes differentiate in association with the glands of Zeis in the eyelids during the invagination of ectoderm to form pilosebaceous units. Distichiasis results from maldevelopment in which cilia formation occurs in association with Meibomian glands. Treatment is directed toward protecting the corneal epithelium. If the lashes do not touch the epithelium, no treatment is required. Electrolysis, cryotherapy, or a surgical eyelid-splitting procedure to remove the hair follicles may be performed when the lashes contact the corneal epithelial surface.
Conjunctiva
Many genetic disorders manifest themselves in the conjunctiva. Patients with ataxia telangiectasia (OMIM 208900: progressive cerebellar ataxia of childhood, oculomotor apraxia, absent optokinetic nystagmus, oculocutaneous telangiectases, immune defects, and malignancy predisposition) have significant dilatation and tortuosity of the conjunctival vessels. A characteristic oculomotor apraxia, that is, difficulty in the initiation of voluntary eye movements, frequently precedes the development of the telangiectases. The ataxia telangiectasia mutated gene on chromosome 11q23.3 (121) encodes a large protein kinase that regulates the cell cycle (122,123).
Congenital or early adult pterygium of the conjunctiva and cornea (OMIM 178000) is inherited as a dominant trait with 70% penetrance (124,125).
Pingueculae are evident in all forms of Gaucher disease (OMIM 230800, 230900, and 231000), one of the glycogen storage disorders (126).
Cornea
Developmental Corneal Abnormalities
Developmental abnormalities of the cornea include cornea plana, sclerocornea, microcornea, megalocornea, and keratoconus.
Cornea plana (CNA1; OMIM 121400, and CNA2; OMIM 217300) is a condition in which the cornea is flat, with a radius of curvature less than 43 D. A cornea with the same radius of curvature as the adjacent sclera is pathognomonic. This condition results from a failure of formation of the limbus by the second wave of neural crest cells during development. Cornea plana maybe associated with microcornea or sclerocornea, as well as cataracts, anterior or posterior colobomata, and Ehlers-Danlos syndrome. Glaucoma may develop as a result of angle abnormalities (open-angle glaucoma) or due to a morphologically shallow anterior chamber (angle-closure glaucoma). Cornea plana has two subtypes distinguished by their inheritance pattern and severity. CNA1 is autosomal dominant, whereas CNA2 is autosomal recessive and the more severe of the two. Keratocan (KERA) mutations are associated with both types, although none were found in the original CNA1 families (127,128). KERA codes for a keratin sulfate proteoglycan and is important for the development and maintenance of corneal transparency and structure. KERA has restricted expression in early neural crest development and later expression in corneal stromal cells. Treatment includes neutralization of refractive errors and glaucoma management. If central clarity is lost, penetrating keratoplasty may be considered.
Sclerocornea (OMIM 269400) is also characterized by a flat cornea; however, it differs from cornea plana by the partial or complete loss of transparency of the cornea. The limbus may be ill-defined, and scleral, episcleral, and conjunctival vessels extend across the cornea. Half of sclerocornea cases are sporadic, while the other half are either dominant or recessive (129).
Microcornea (OMIM 116150) is a condition in which the cornea is clear and of normal thickness; however, it measures less than 10 mm in diameter (9 mm in a newborn). It
is thought to result from either fetal arrest of growth of the cornea or from overgrowth of the anterior tips of the optic cup leaving less space for the cornea to develop. It exists in both autosomal dominant and recessive forms, with the former occurring more commonly. Treatment includes correction of refractive errors and monitoring for the development of glaucoma due to shallow chambers or angle abnormalities.
is thought to result from either fetal arrest of growth of the cornea or from overgrowth of the anterior tips of the optic cup leaving less space for the cornea to develop. It exists in both autosomal dominant and recessive forms, with the former occurring more commonly. Treatment includes correction of refractive errors and monitoring for the development of glaucoma due to shallow chambers or angle abnormalities.
Megalocornea (OMIM 249300) is a condition in which the corneal diameter is large (13.0 to 16.5 mm), but the cornea is clear and of normal thickness. The enlargement is not a result of congenital glaucoma. This condition represents either a failure of the anterior tips of the optic cup to close, allowing more room for the cornea to grow, or an overgrowth of the cornea in relation to the rest of the eye. It may be associated with a number of ocular abnormalities including miosis, microcoria, goniodysgenesis, cataract, ectopia lentis, central cloudy dystrophy of Francois, and glaucoma. Furthermore, it is associated systemically with disorders of collagen synthesis. It is usually X-linked recessive and located in the Xq21.3-q22 region. There are reports of sporadic, autosomal dominant and recessive cases. A syndromic form of megalocornea, termed megalocornea and mental retardation syndrome (OMIM 249310), was first reported by Neuhauser and associates (130). Children are hypotonic and show mild frontal bossing, antimongoloid slant of the eyes, epicanthal folds, and broad nasal base. Iris hypoplasia may accompany the megalocornea.
Keratoconus (OMIM 148300) can be sporadic or autosomal dominant. The onset is around puberty with a progressive ectatic dystrophy leading to corneal thinning, with induced irregular myopic astigmatism which may be markedly asymmetrical. In advanced cases, anterior corneal scarring is present and hydrops may occur when Descemet’s membrane ruptures with subsequent epithelial and stromal edema. Either circumstance may require a corneal graft to attain visual clarity. Keratoconus has been associated with several chromosomal anomalies including trisomy 21, Turner syndrome, ring chromosome 13, and chromosomal 7;11 translocation; connective-tissue disorders such as Ehlers-Danlos, Marfan, and osteogenesis imperfecta syndromes; mitral valve prolapse; Leber congenital amaurosis; and atopy (131,132). Mutations in the VSX-1 transcription factor gene were identified in 4.7% of patients with isolated keratoconus. This gene also plays a role in posterior polymorphous dystrophy (see below) (133).
Corneal Dystrophies
Hereditary dystrophies affecting all layers of the cornea are numerous; most are rare, and ophthalmologic evaluation is required to distinguish among them. Most of the corneal dystrophies are of Mendelian inheritance with some phenotype diversity and a variable degree of penetrance. The dystrophies involving enzymatic processes tend to be of autosomal recessive inheritance. Corneal clouding may be present at birth or is acquired (Fig. 1.12). Presenting signs and symptoms of the corneal dystrophies include cloudiness of the cornea, nystagmus due to poor vision, and photophobia. Corneal transplantation may be performed if vision is seriously reduced.
 FIGURE 1.12. Opacification of corneal stroma in a 10-year-old patient with Maroteaux-Lamy syndrome (mucopolysaccharidosis VI). |
Meesmann corneal dystrophy (OMIM 122100) has an autosomal dominant inheritance with an onset in early childhood (at approximately 12 months). Formed intraepithelial vesicles and microcysts, which contain periodic acid Schiff-positive “peculiar substances” suggestive of keratin, increase in number throughout life. The symptoms are variable, ranging from asymptomatic to pain and lacrimation associated with corneal erosions. Mutations have been identified in both keratin 12 (OMIM 601687) on chromosome 12q12-q13 and keratin 3 (OMIM 148043) on chromosome 17q12. Both genes, which are expressed in the anterior corneal epithelium, contain a highly conserved helix boundary motif, which plays a role in corneal structural integrity and keratinocyte filament assembly (134).
Defects in the human transforming growth factor β-induced gene (TGFβI or βIGH3) (135) are associated with multiple “classic” corneal dystrophies: lattice (OMIM 122200), non-classic LCDIIIa, intermediate-type LCDI/LCDIII, and LCD-deep dystrophies; granular (OMIM 121900) and nonclassic granular dystrophies GCDII and GCDIII; Reis-Bucklers (OMIM 608470); Thiel-Behnke dystrophy (OMIM 602082); and Avellino corneal dystrophy. All of the corneal dystrophies show autosomal dominant hereditary transmission with variable penetrance and map to chromosome 5q31 (136,137,138,139). TGFβI is expressed in keratocytes and encodes for keratoepithelin, a highly conserved 683-amino acid protein. This protein contains an N-terminal secretory signal, four domains of internal homology, and an arginine-glycine-aspartame (RGD) motif at the carboxy terminus, which is found in many extracellular matrix proteins. The RGD motif modulates cell adhesion and acts as a recognition sequence for integrin binding. Gene mutations result in progressive accumulation of keratoepithelin corneal deposits.
Aggregation of abnormal isoforms of keratoepithelin is associated with amyloid or other nonfibrillar deposits depending on the type of mutation (140).
Aggregation of abnormal isoforms of keratoepithelin is associated with amyloid or other nonfibrillar deposits depending on the type of mutation (140).
The different subtypes of macular dystrophy, MCDI (OMIM 217800), MCDIa, and MCDII, are genetically and biochemically determined. The dystrophy is inherited in an autosomal recessive fashion, with onset in the first decade of life. Early on, the fine opacities have indistinct edges which start axially in the superficial stroma. The intervening stroma has a ground-glass appearance. Later, the opacities extend peripherally and into the deep stroma. The characteristic accumulation of glycosaminoglycans stains with Alcian blue and colloidal iron. MCDI is characterized by the absence of keratin chain sulfation (KCS) in cornea and cartilage and no appreciable serum KCS. In MCDII, serum and corneal keratin sulfate are detectable. MCD was mapped to chromosome 16q22 and mutations were noted in CHST6 (OMIM 603797). CHST6 encodes an enzyme, carbohydrate sulfotransferase, which is expressed in the cornea, spine, and trachea. The gene product initiates sulfation of keratin sulfate in the cornea. Mutations in this gene result in an inactive enzyme with the synthesis and secretion of proteoglycans (corneal structural genes) substituted with polylactosamine instead of keratin sulfate (141,142,143). Gelatinous drop-like corneal dystrophy (GDLD; OMIM 204870) is an autosomal recessive disorder with clinical onset in the first decade of flat subepithelial nodular deposits similar to early band keratopathy. There is a gradual increase in number and depth of deposits which eventually become raised as yellow-gray gelatinous masses with a mulberry configuration and surrounding dense subepithelial opacities. Recurrent lamellar keratoplasty or penetrating keratoplasty may be required. The gene, M1S1, maps to chromosome 1p31 and encodes for a gastrointestinal tumor-associated antigen. The abnormal M1S1 gene product may affect epithelial cell junctions, resulting in increased cell permeability in GDLD corneas (144,145).
Posterior polymorphous corneal dystrophy (PPCD; OMIM 122000, 120252, and 605020) is an autosomal dominant disorder which shows variable expression and variable age of onset. Although it is usually a disease of adulthood, PPCD can be severe and present at birth. The disorder manifests as variable degrees of vesicular endothelial lesions with or without basement membrane thickening. This may be localized or diffuse, and may be associated with corneal edema. There is an increased risk for glaucoma and keratoconus. The abnormal anterior banded layer of Descemet’s membrane is lined posteriorly by an abnormal posterior collagenous layer. PPCD is genetically heterogeneous with mutations identified in the transcription factor VSX1 (chromosome 20p11.2-20q11.2) and the alpha 2 subunit of type VIII collagen COL8A2 (chromosome 1p34.3-p32). PPCD is an allelic variant of keratoconus (OMIM 148300) and Fuchs’ endothelial dystrophy (FECD; OMIM 136800), as VSX1 appears to play a role in approximately 9% of PPCD cases and 4.5% of keratoconus cases, and COL8A2 appears to play a role in approximately 6% of PPCD cases and 3.4% of FECD cases, respectively (133,146).
Congenital hereditary endothelial dystrophy (CHED1; OMIM 121700) is of autosomal dominant inheritance with onset at birth or in the first few months of life up to 8 years of age. CHED2 (OMIM 217700) is of autosomal recessive inheritance with an early onset of signs and symptoms at birth or within the first few weeks of life. The cornea has a ground-glass appearance, and the corneal epithelium may be roughened. There is no guttatae and the corneal sensitivity is normal. The decrease in vision is moderate to severe, and nystagmus is uncommon. The basement membrane is more thickened in CHED2. CHED is genetically heterogeneous, with CHED1 linked to a 2.7-cM locus at chromosome 20p11.2-20q11.2, and CHED2 linked to a chromosome 20p13 locus. No genes have been identified as of yet (147,148).
Anterior Segment Dysgenesis
Malformations of the iris and anterior chamber angle may involve the region responsible for aqueous humor outflow and predispose patients to glaucoma.
As an isolated ocular malformation, aniridia (OMIM 106200) is an autosomal dominant disorder (Fig. 1.3). Visual impairment, which is unrelated to the degree of iris hypoplasia, is caused either by glaucoma or the frequent concomitant malformations of macular or optic nerve hypoplasia. Clinical expressivity can be variable within and among families (149,150). There are multiple reports of aniridia, developmental delays, genitourinary malformations, and Wilms’ tumor associated with a deletion of the short arm of chromosome 11 (151,152,153,154); one patient had aniridia and Wilms’ tumor without a microscopically detectable deletion (155) (see section “Chromosomal Rearrangements”). Autosomal dominant aniridia is due to a mutation in the PAX6 gene (156,157). Mutations of this gene can cause other disorders, including congenital cataracts, anophthalmia and central nervous system defects (158), Peters’ anomaly (159), and keratitis (160).
Axenfeld-Rieger syndrome (OMIM 602482 and 180500) is a spectrum of anterior segment and systemic structural change combinations which may be characterized by a prominent Schwalbe line, iris strands to the cornea, iris hypoplasia, dental abnormalities, characteristic facies, and umbilic defects; it is generally inherited in an autosomal dominant fashion. Associated ocular abnormalities include glaucoma, corectopia, iris pigment epithelial defects, microcornea, corneal opacities, and cataracts (161,162,163,164,165,166). Uncommonly, chromosomal rearrangements may cause Rieger syndrome, in which case there is usually developmental delay (167,168,169,170,171,172,173,174). Mutations of the eye developmental genes FOXC1, a forkhead box transcription factor, and PITX2, paired-like class of homeobox transcription factors, cause Axenfeld-Rieger syndrome (175,176,177,178,179). FoxC1 knockout mice also have anterior segment abnormalities that are similar to those reported in humans. The penetrance of
the clinical phenotype varies with the genetic background, which indicates the influence of modulator genes.
the clinical phenotype varies with the genetic background, which indicates the influence of modulator genes.
In humans, mutations in PITX3 cause anterior segment mesenchymal dysgenesis (180). In the mouse embryo, Pitx3 is first expressed in the developing lens, initially in the lens vesicle and later in the anterior epithelium and lens equator. Deletions in the Pitx3 promoter, which abrogate Pitx3 expression in the eye, cause the phenotype of the aphakia (ak) mouse mutant, which lacks lenses and pupils (181).
Peters’ anomaly may result from incomplete separation of the cornea and lens during embryogenesis; although evidence supports an autosomal recessive form of inheritance, chromosomal and nongenetic forms may exist (182,183,184,185). Mutations in the eye development genes PAX6, PITX2, FOXC1, and CY1B1 have all been associated with Peters’ anomaly (159,178,186,187).
Glaucoma
Glaucoma describes a heterogeneous group of optic neuropathies that lead to optic nerve cell damage, visual field loss, and permanent visual acuity deficits. It is the second most prevalent cause of bilateral blindness in the Western world, and it affects more than 60 million people worldwide. The hereditary forms of glaucoma are genetically heterogeneous. At least eight loci have been linked to glaucoma (GLC1A-F, GLC3A/B), and three genes have been identified to date: MYOC, CYP1B1, and OPTN.
Primary congenital glaucoma (PCG; OMIM 231300) is defined by the onset before 3 years of age. It is often characterized by buphthalmos, or an enlarged eye, as a result of increased intraocular pressure during intrauterine life or infancy when the elasticity of the scleral wall and cornea are greatest. Structurally, these eyes have a normal Schlemm’s canal and normal episcleral veins; however, the trabecular meshwork is thought to be abnormal, impeding the drainage of aqueous humor from the anterior chamber. Infants with congenital glaucoma present with the classic triad of epiphora, photophobia, and blepharospasm. On examination, their corneal diameters may be markedly enlarged (greater than 12 mm during the first year of life), and the eye may be buphthalmic. Corneal edema may develop due to increased intraocular pressure, and acute stretching of the cornea may lead to Haab’s striae (tears in Descemet’s membrane). Inheritance is thought to be recessive with incomplete penetrance, and two loci have been mapped: one to chromosome 2p22-p21 (188,189) and the second to chromosome 1p36.2-p36.1 (190). Mutations of the gene CYP1B1 (OMIM 601771) on chromosome 2p21 are associated with PCG (191). The gene encodes a 543amino acid drug-metabolizing enzyme of the cytochrome P450 gene superfamily, subfamily I, which is a monooxygenase that is capable of metabolizing various endogenous and exogenous substrates, such as steroids and retinoids. It is expressed in the iris, trabecular meshwork, and ciliary body. Congenital glaucoma has also been reported to occur in association with deletions of chromosomes 10, 13, and 18; a pericentric inversion of chromosome 11; partial trisomy of chromosomes 3 and 14; and trisomies 13, 18, and 21 (192,193). Treatment is primarily surgical for congenital glaucoma, via a trabeculotomy or goniotomy.
Juvenile-onset open-angle glaucoma (JOAG; OMIM 137750) is defined as glaucoma acquired after birth and thus unaccompanied by buphthalmos. It is dominantly inherited and typically has its onset during the second or third decade of life. There is an association with myopia reported to be as high as 87% (194). Mutations in the myocilin gene (MYOC; also known as trabecular meshwork-inducible glucocorticoid response gene; OMIM 601652) are associated with autosomal dominant JOAG (33% of patients) and with primary open-angle glaucoma (POAG; OMIM 137760) (2% to 4% of patients) (195,196). MYOC has been mapped to a locus (GLC1A) on chromosome 1q24.3-q25.2. MYOC is expressed in almost every ocular tissue, including the optic nerve (197); however, despite considerable effort, the function of MYOC remains obscure.
POAG is a chronic, progressive optic neuropathy characterized by cupping of the optic nerve head and is associated with visual field loss. Intraocular pressure may be elevated. It is insidious in onset and often progresses without symptoms since central visual acuity is relatively unaffected until late in the disease. Inheritance is thought to be multifactorial. In addition to the myocilin gene-associated GLC1A locus on chromosome 1q23-24, POAG has been mapped to several other loci: GLC1B on chromosome 2cenq13, GLC1C on chromosome 3q21-24, GLC1D on chromosome 8q23, and GLC1F on chromosome 7q35-q36. Recently, mutations in the optineurin gene (OPTN), a locus previously designated GLC1E on chromosome 10p14-p15, have been demonstrated in 16.7% of patients with POAG (198). The pathogenic mechanism for glaucoma development due to mutations in OPTN is not fully understood—it is speculated that OPTN is operating through an apoptosis (cell death) pathway, playing a neuroprotective role in the eye and optic nerve (198).
Pigment dispersion syndrome (OMIM 600510) is a form of open-angle glaucoma which is characterized by pigment deposition on the corneal endothelium (Krukenberg spindle), trabecular meshwork, and lens periphery. Spokelike loss of the iris pigment epithelium occurs, resulting in characteristic transillumination defects in the iris midperiphery. This loss of pigment is thought to be due to direct contact between the zonules and iris. Affected individuals are usually myopic men aged 20-50 years. Fluctuations in intraocular pressure are usually wide, and patients are symptomatic with headaches, haloes, intermittent visual blurring, and pain. Medications are often helpful in controlling intraocular pressure; laser iridotomies are often attempted in cases of “reverse pupillary block,” and laser trabeculoplasty and filtering surgery are useful in treatment. Inheritance is autosomal dominant (199), and the gene has been mapped to chromosome 7q35-q36 (200).
Lens
Abnormalities of the lens of the eye may be divided into two broad categories: dislocations and opacities. A lens is subluxed if it is not in its proper anatomic location but still retains some zonular attachments to the ciliary body; in a complete dislocation, the lens floats free in the eye. Lens opacities, or cataracts, may occur in different layers of the lens and with varying severity.
The causes of subluxed and dislocated lenses are numerous. Dislocation or subluxation of the lens is a common manifestation of both Marfan syndrome (OMIM 154700) (Fig. 1.13) (201) and homocystinuria (OMIM 236200). Common manifestations in Marfan syndrome include lens subluxation superotemporally, dilatation of the aortic root, aneurysm of the ascending aorta, and skeletal abnormalities such as pectus excavatum, kyphoscoliosis, and an upper segment: lower segment ratio two standard deviations below the mean for age. Pneumothorax, pes planus, dural ectasia, joint laxity, arachnodactyly, and mitral valve prolapse may also occur. The diagnostic criteria require involvement of three systems with at least two major manifestations. As mentioned previously, the basis of Marfan syndrome is a mutation of the fibrillin gene, which maps to chromosome 15q15-q21.1 (202,203,204,205,206,207,208).
Homocystinuria is characterized by a deficiency of the enzyme cystathione-β-synthase, resulting in accumulation of homocysteine. Untreated patients present with mental retardation, unusually tall stature, coarse fair hair, cardiac murmurs, and thromboembolic diathesis. These patients have an increased anesthetic risk due to their tendency to develop thrombotic vascular occlusions. Ocular findings include blue irides and progressive myopia, which may be the first sign of lens dislocation. All untreated patients develop inferior or inferonasal dislocation of lenses bilaterally due to deficient zonular fibers. Diagnosis is confirmed by measurement of the serum homocysteine level. Surgery should be avoided if possible, and treatment consists of dietary management (low methionine and high cysteine) and supplementation of vitamin B6. The gene for cystathione-β-synthase has been mapped to 21q22.3 (209), and it is transmitted in an autosomal recessive fashion.
 FIGURE 1.13. Superior dislocation of the lens in a young girl with Marfan syndrome. |
Ectopia lentis et pupillae (OMIM 225200), an autosomal recessive disorder, is characterized by an eccentric pupil and subluxed lens (usually the lens is dislocated in a direction opposite the pupil) and is slowly progressive (210). Isolated lens subluxation or dislocation is rare, and most reports antedate careful definition of the Marfan syndrome and homocystinuria.
Isolated hereditary cataracts are usually transmitted in an autosomal dominant pattern, although autosomal recessive and X-linked forms have been reported. They may be present at birth or develop over time. Although the severity may vary within a family, the position and patterns are generally consistent. This variability within the same family suggests the importance of additional genes modifying the expression of the primary mutation. Conversely, cataracts with similar or identical clinical presentations can result from mutations in quite different genes. Currently, 27 isolated or primary cataract loci have been identified by linkage analysis or mutational screening, and 13 are associated with specific gene defects. For some α- and β-crystallin mutations, inherited congenital cataracts are associated with microcornea and even microphakia. There are currently no identified developmental lesions causing isolated cataracts. Of those families for whom the mutant gene is known, about half have mutations in crystallins (structural components of the lens nucleus), about one-fourth have mutations in connexins (constituents of gap junctions on which the avascular lens depends for nutrition and intercellular communication), and the remainder are evenly split between aquaporin 0 (an enzyme involved in water-channel activity) and the gene for the beaded filament protein BFSP2 (structural filament unique to the lens fiber cells that combines with β-crystallin) (158,211,212,213,214,215,216,217,218,219,220,221,222,223).
Cataracts are known to occur in association with a large number of metabolic diseases and genetic syndromes. Nance-Horan syndrome (OMIM 302350) is an X-linked recessive congenital cataract-dental disorder associated with microcornea, anteverted and simplex pinnae, brachymetacarpalia, and various dental anomalies; carriers exhibit distinctive sutural opacities (26,27). An isolated X-linked cataract has recently been mapped to chromosome Xp and is possibly allelic with the Nance-Horan syndrome (224).
Galactosemia (OMIM 230400) is an autosomal recessive disorder characterized by the inability to convert galactose to glucose, resulting in the accumulation of galactose and its conversion to galactitol. Classic galactosemia, which is the most common of three forms of the disease, involves a defect in galactose-1-phosphate uridyl transferase (GALT) and results in cataract formation during the first few weeks of life in 75% of untreated patients. Two other, less severe forms of the disease involve defects in two other enzymes, galactokinase and UDP-galactose-4-epimerase. Patients
with classic galactosemia present with malnutrition, hepatomegaly, jaundice, and mental deficiency within the first few weeks of life. The cataract has a very characteristic “oil droplet” appearance on retroillumination due to accumulation of galactose and galactitol within the lens cells causing increased intracellular osmotic pressure and fluid influx into the lens. Diagnosis can be confirmed by testing the urine for reducing substances and demonstrating the presence of the nonglucose reducing substance, galactose. Treatment includes elimination of milk and milk products from the diet. GALT



with classic galactosemia present with malnutrition, hepatomegaly, jaundice, and mental deficiency within the first few weeks of life. The cataract has a very characteristic “oil droplet” appearance on retroillumination due to accumulation of galactose and galactitol within the lens cells causing increased intracellular osmotic pressure and fluid influx into the lens. Diagnosis can be confirmed by testing the urine for reducing substances and demonstrating the presence of the nonglucose reducing substance, galactose. Treatment includes elimination of milk and milk products from the diet. GALT
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
