2
Genetic Predictors of Cochlear Implant Outcome
Andrew K. Patel and Anil K. Lalwani
Congenital or acquired severe to profound loss sustained prior to the development of language is estimated to occur between 0.5 and 4.0 per 1000 births (Morton, 1991). The most common cause of childhood deafness is genetic, accounting for approximately one half to two thirds of all cases (Marazita et al, 1993). The remainder of cases of childhood deafness is due to nongenetic or environmental causes; however, it is likely that genetic factors contribute to many of these forms of congenital deafness.
Several hundred genes are thought to cause hereditary hearing loss and deafness (Gorlin et al, 1995). A large number of genetic hearing loss cases are single-gene mutations, and this proportion has continued to increases as public health improves, leading to a decrease in the incidence of hearing loss resulting from infection. Hereditary hearing loss is classified into syndromic and nonsyndromic forms. The syndromic forms, in which hearing loss is associated with a variety of other anomalies, account for 30% of genetic deafness; several hundred syndromes associated with hearing loss have been described (Gorlin et al, 1995). Among hereditary nonsyndromic deafness, autosomal recessive forms predominate (accounting for ˜85% of cases), followed by autosomal dominant, X-linked, and mitochondrial forms (Petit, 1996; Van Camp, 2005). The last few years have seen major advances in the elucidation of the genetic causes of childhood-onset hearing impairment. Since 1992, nearly 120 loci for nonsyndromic deafness have been mapped to human chromosomes and 40 responsible genes have been identified—quite a remarkable feat given that only one deafness gene had been mapped in 1992 (Petit, 1996; Van Camp et al, 1997; Snoeckx et al, 2005).
♦ Modes of Inheritance in Nonsyndromic Deafness
Recessive Hearing Impairment
Recessive hearing impairment is typified by profound hearing loss at birth and accounts for nearly 85% of genetic hearing impairment. An autosomal recessive trait is characterized by having two unaffected parents who are heterozygous carriers for mutant forms of the gene in question but in whom the phenotypic expression of the mutant allele is masked by the normal allele (Fig. 2–1). These heterozygous parents (A/a) can each generate two types of gametes, one carrying the mutant copy of the gene (a) and the other having a normal copy of the gene (A). Of the four possible combinations of these two gamete types from each of the parents, only the offspring that inherits both mutant copies (a/a) will exhibit the trait. Of the three remaining possibilities, all will have a normal hearing phenotype but two of the three will be heterozygous carriers for the mutant form of the gene, similar to the carrier parents. Marriage among close relatives greatly increases the potential to produce a child affected by a recessive disorder.
Autosomal recessive deafness is extremely heterogeneous. Over 59 separate nonsyndromic recessive loci (noted as DFNBX, where X represents the order in which it was identified) have been reported, and already at least 21 of these genes have been identified. The most important of these is GJB2 encoding for a gap junction protein connexin 26. Mutations in this gene, which facilitates transport of small solutes across cells, are responsible for half of childhood recessive deafness. Other recessive genes identified thus far encode proteins involved in the structure or maintenance of the inner ear, particularly of hair cells, or in the maintenance ionic balances within the inner ear.
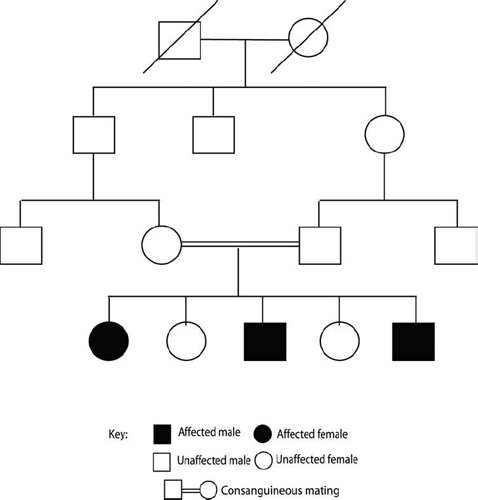
Figure 2–1 Autosomal recessive inheritance. Pedigree demonstrates typical recessive inheritance where affected individuals are primarily seen in a single generation (horizontal transmission). To be affected, the child needs to inherit the mutated gene from each of its unaffected parents. Risk of recessive diseases increases in consanguineous marriages, as shown here among two cousins, because they already share similar genetic information. Examples of recessive disorders include GJB2-related deafness (DFNB1), Pendred syndrome, and Usher syndrome.
Autosomal Dominant Hearing Impairment
Autosomal dominant hearing impairment is characterized by delayed-onset, progressive hearing loss and accounts for 10 to 15% of genetic hearing impairment. For autosomal dominant disorders, the transmission of a rare allele of a gene by a single heterozygous parent is sufficient to generate an affected child (Fig. 2–2A). A heterozygous parent can produce two types of gametes. One gamete carries the mutant form of the gene of interest, and the other carries the normal form. Each of these gametes then has an equal chance of being utilized in the formation of a zygote. Thus the chance that an offspring of an autosomal dominant affected parent will be affected is 50%. Equal numbers of affected males and females are expected for an autosomal dominant trait and roughly half of the offspring of an affected individual will be affected.
Autosomal dominant traits often exhibit incomplete penetrance and variable expressivity. Variable expressivity refers to the differences in the observed effects of a given allele in related and unrelated individuals. Variable expression of different aspects of syndromes including hearing loss is common. The range of variation observed in dominant disorders is generally wider than in recessive disorders. Incomplete penetrance is an extreme form of variable expressivity and is characterized by the total absence of expression in persons known to carry the mutant allele (Fig. 2–2B). Variation in the age of onset for symptoms associated with a genetic disorder is common for traits that are not expressed at birth or prenatally. If the mutant phenotype is always expressed in individuals carrying the disease allele, then its penetrance is said to be complete, otherwise it is incomplete. Complete penetrance of the dominant allele results in the expression of the disease phenotype in all carriers of that allele without skipping generations.
Similar to autosomal recessive nonsyndromic hearing impairment, autosomal dominant nonsyndromic hearing impairment is genetically heterogeneous. To date, 54 chromosomal regions have been shown to contain a gene that is involved in an autosomal dominant type of nonsyndromic hearing impairment, the so-called DFNA loci. Twenty-one of the corresponding genes have been identified, including channel and gap junction proteins, unconventional myosins, transcription factors, extracellular matrix proteins, and genes with an unknown function. These gene identifications are increasing knowledge of the molecular basis of hearing, and potentially may lead to new therapeutic measures for hearing impairment in the future.
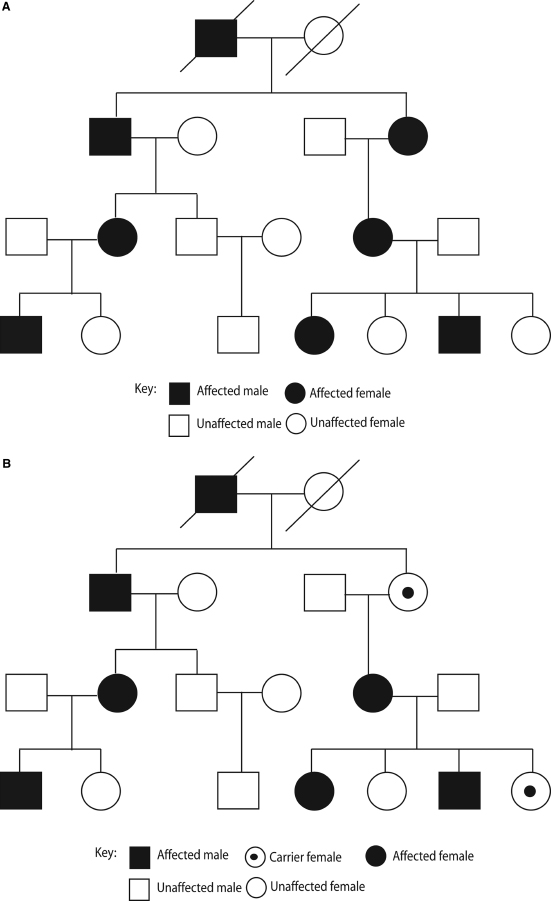
Figure 2–2 Autosomal dominant inheritance. (A) Pedigree showing typical autosomal dominant inheritance pattern where affected individual are seen in multiple generations (vertical transmission). To be affected, an offspring needs to inherit only a single copy of the mutated gene from either parent. This pedigree demonstrates complete penetrance as every individual with the affected gene is affected. (B) In contrast, this pedigree demonstrates incomplete penetrance of a dominant trait: every individual who harbors the mutated gene does not have the associated disease. Examples of dominant diseases include Waardenburg syndrome, neurofibromatosis type 2, and Alport syndrome.
X-linked Hearing Impairment
X-chromosomal inherited nonsyndromic hearing impairment accounts for about 2 to 3% of all hearing impairment and is therefore less common than autosomal hereditary hearing impairment (Fig. 2–3). The X-linked or DFN phenotype displays a high grade of clinical and genetic heterogeneity. In humans, females have 22 pairs of autosomes and a pair of X chromosomes (46, XX); males have 22 autosomes, one X chromosome, and one Y chromosome (46, XY). Accordingly, males always receive their Y chromosome from their father and their X chromosome from their mother, whereas females receive one of their X chromosomes from each of their two parents. Men are affected more often and, because of hemizygosity, affected males usually show a more severe, distinctive hearing impairment phenotype. Diseases that are rarely expressed clinically in heterozygous females are called X-linked recessive; if they commonly present in females, it is designated X-linked dominant. Currently, five nonsyndromic loci and one syndromic locus have been identified and two genes have been cloned. Stapes gusher syndrome is an X-linked disorder for which the responsible gene, POU3F4, has been identified. Patients often show a prelingual onset of hearing impairment.
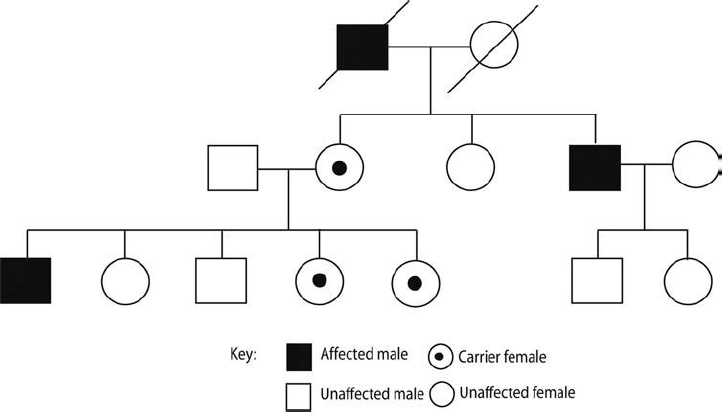
Figure 2–3 X-linked inheritance. Pedigree showing a typical X-linked inheritance with affected males and unaffected carrier females. Notice that fathers cannot pass along the X chromosome harboring the mutation to their sons as they contribute the Y chromosome; further, the daughters are only carriers as they inherit a normal X chromosome from their mother. In contrast, carrier mothers have affected and unaffected sons and carrier daughters. Examples of X-linked disorders include stapes gusher syndrome, Duchenne muscular dystrophy, and color blindness.
Mitochondrial Mutations and Hearing Impairment
Not all genes are equally inherited from both parents. The extranuclear genome is inherited solely through the mother. Male mitochondria are not contributed to newly formed zygotes. This inheritance pattern gives rise to pedigrees in which all the children of an affected mother may be affected and none of the children of an affected father will be affected (Fig. 2–4). In practice, the expression of mitochondrially inherited disorders is often variable and may be incompletely penetrant.
The last decade has seen the identification of several mitochondrial DNA mutations associated with hearing loss. Because the only known function of the human mitochondrial chromosome is to participate in the production of chemical energy through oxidative phosphorylation, it was not unexpected that mitochondrial mutations interfering with energy production could cause systemic neuromuscular disorders, which have as one of their features hearing impairment. Surprisingly, however, inherited mitochondrial mutations have also been found to be a cause of nonsyndromic hearing loss and to predispose to aminoglycoside-induced hearing loss, whereas acquired mitochondrial mutations have been proposed as one of the causes of presbycusis. Clinical expression of these mitochondrial mutations is dependent on environmental exposures and nuclear-encoded modifier genes. Emphasis on preventive or therapeutic strategies requires identification and avoidance of the environmental exposures, and the identification of the nuclear-encoded modifier genes.
♦ The Impact of Cochlear Implantation on Hereditary Hearing Impairment
Cochlear implantation has been used to restore the sense of audition to deaf children since the mid-1980s, when the first single-channel devices followed by multichannel devices were permanently implanted. Since that time, significant advances have been made in both device design and implantation techniques. These have allowed the technology to be more broadly and successfully applied. Candidacy guidelines based in part on consistent, reliable documentation of postoperative performance have evolved greatly since the mid-1980s. Patient selection has emerged as one of the most important determinants of successful outcome after pediatric cochlear implantation (Daya et al, 1999).
To determine suitability for cochlear implantation, fulfillment of several criteria is necessary. These include confirmation that there is a profound sensorineural hearing loss (SNHL), an absence of medical contraindications to implantation, and the presence of an implantable cochlea. Further evaluation of speech and language development, educational setting, home environment, developmental milestones, and other handicaps facilitates determining the degree and type of rehabilitation appropriate for the implant candidate. Finally, assessment of factors such as age at onset of deafness, duration of deafness, and speech perception performance may provide some indication of expected outcome. Assessment therefore requires a multidisciplinary approach. Many centers have developed a structured approach to evaluate potential candidates to select those who would benefit the most from implantation.
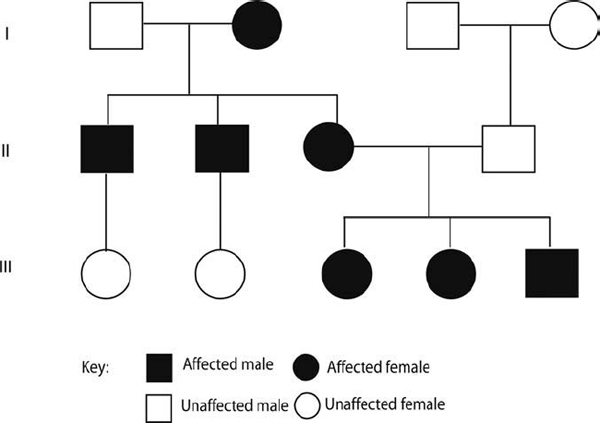
Figure 2–4 Mitochondrial inheritance. Pedigree showing mitochondrial inheritance in a three-generation (I, II, III) family. As the mitochondrial DNA is only present in the egg and not the sperm, the mitochondrial disease can only be passed on by an affected mother. An example of mitochondrial disease includes hearing loss due to exaggerated sensitivity to aminoglycosides associated with A1555G mutation in 12S ribosomal RNA.
Cochlear implantation (CI) is an effective but controversial intervention that poses multiple challenges to both purchasers and providers of health care. Although offering a potentially life-transforming benefit for patients, the relative expense for the procedure and the subsequent rehabilitation may be high. Cochlear implant manufacturers seek to expand the market by promoting relaxations of cochlear implant candidate criteria. Although more than 90% of congenitally deaf children are born to hearing parents who wish to communicate with their children by speaking rather than signing, the question arises whether CI is sufficiently effective and cost-effective to justify elective surgery on a young child (Lane & Bahan, 1998); recent long-term follow-up in children implanted under the age 2 strongly suggests that CI is effective in rehabilitation of the deaf. Finally, CI is a low-volume intervention at most hospital centers, with variable outcomes whose overall effectiveness is difficult to judge from outcomes obtained in any individual hospital.
♦ Prediction of Successful Outcomes with Cochlear Implantation
It is strongly desirable to identify factors that predict cochlear implantation outcome. At this time, no single test can predict which patients will achieve success with CI. Evidence supporting the efficacy of cochlear implants in sensorineural deafness exists primarily in the form of data from several prospective and retrospective case series, comparative case series, and matched-pair case series. The reported benefit in children has varied widely across individual users. Therefore, although one can inform the patient or the patient’s parents that most do well with implants, it is not possible to predict the outcome for any given patient.
With the advances in our understanding of genetics of deafness, recent work has focused on the ability of genetic testing to predict CI outcome. Currently, genetic testing is available for many different types of syndromic and nonsyndromic deafness, though most are only available on a research basis. However, DNA-based testing can be performed to diagnose Pendred syndrome (SLC26A4 gene), Usher syndrome type IIA (USH2A gene), branchio-oto-renal (BOR) syndrome (EYA1 gene), Mohr-Tranebjaerg syndrome (deafness-dystonia-optic atrophy syndrome; TIMM8A gene), one mutation in USH3A, DFNB1 (GJB2 gene), DFN3 (POU3F4 gene), DFNB4 (SLC26A4 gene), and DFNA6/14 (WFS1 gene) (Van Camp, 2005). Diagnostic testing for mutations in the GJB2 gene causing deafness (which encodes the connexin 26 protein) and in the GJB6 gene (which encodes the connexin 30 protein) also plays a prominent role in diagnosis and genetic counseling due to the high proportion of nonsyndromic hearing loss caused by these mutations.
♦ Evaluation of Specific Causes of Hereditary Hearing Impairment and Cochlear Implant Outcome
GJB2-Related Deafness and Cochlear Implant Outcome
After a newborn screening test indicates hearing loss, the neonate is referred for evaluation by a multidisciplinary team experienced in the evaluation of neonatal deafness. Hearing loss is categorized by the portion of the hearing system affected (conductive, sensorineural, or mixed); whether it is primarily due to a genetic cause or environmental one (hereditary or acquired), and if genetic, whether or not it is a component of a genetic syndrome (syndromic or nonsyndromic). If genetic, 50% are due to DFNB1 mutations. DFNB1 related hearing loss has approximate prevalence in the general population of 14/100,000 (˜1/7,000), with an estimated carrier frequency of 1 in 31 in the general population.
DFNB1-related hearing loss is characterized by congenital (present at birth), nonprogressive sensorineural hearing impairment. Usually, the hearing impairment is severe or severe-to-profound; however, it can range from mild to severe in different families and within a family. Except for the hearing impairment, affected individuals are healthy and enjoy a normal life span. Vestibular function is normal; affected infants and young children do not experience balance problems and learn to sit and walk at age-appropriate times.
DFNB1-related hearing loss is suspected in patients who have congenital, nonprogressive sensorineural hearing impairment that is mild-to-profound by auditory brainstem response (ABR) testing or pure tone audiometry, and no related systemic findings identified by medical history or physical examination. They may also have a family history consistent with autosomal recessive inheritance of hearing loss (for example, an affected sibling). However, the majority of people with autosomal recessive diseases represent the first known case in their family. The diagnosis of DFNB1-related hearing loss is confirmed if the patient has recognized disease-causing mutations in the gene GJB2 (chromosome 13q11–12) that alters the connexin 26 (Cx26) protein. DNA-based testing of the GJB2 gene detects ˜95% of disease-causing mutations. The most common mutation, 35delG, is found in over two thirds of persons with DFNB1.
About 30% of patients with DFNB1 have other identifiable disease-causing mutations in GJB2; more than two dozen other disease-causing mutations have been identified. Available methods of screening for Cx26 mutations have failed to identify disease-causing mutations in some families in whom the diagnosis of DFNB1 has been established by linkage studies; thus, failure to detect a Cx26 mutation does not exclude the diagnosis of DFNB1.
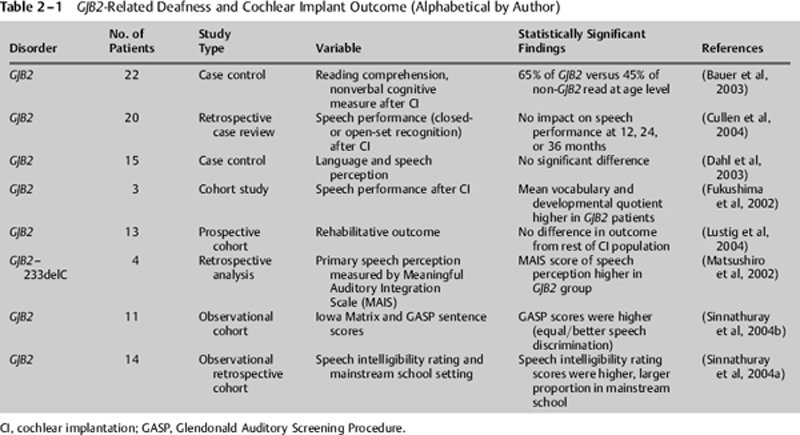
The GJB2 test has prognostic value. Children with profound deafness due to Cx26 mutations have an excellent outcome with cochlear implants (Table 2–1). The isolated insult to the cochlea created by GJB2 allele variants allows for preservation of central cognitive function. Better reading performance is seen in children with GJB2-related deafness compared with children with other nonsyndromic deafness (Bauer et al, 2003). Given that children with GJB2-related deafness do not have other handicaps, improved outcome in this group may reflect the exclusion of children with multiple handicaps in whom the outcome is more heterogeneous. Regardless, excellent outcome in children with GJB2-related deafness is an important piece of information for parents to know as they make decisions regarding rehabilitative intervention in their child.
Syndromic Autosomal Recessive– Related Deafness and Cochlear Implant Outcome
The experience with cochlear implantation in a variety of recessive hearing disorders is summarized in Table 2–2 and is discussed here. Usher syndrome is an autosomal recessive disorder manifested by hearing impairment, retinitis pigmentosa (RP), and variable vestibular deficit, and affects 50% of the deaf-blind patients in the United States. Recent progress in the characterization of the genetics of Usher syndrome has shown that this disorder is phenotypically and genetically complex (Kimberling & Moller, 1995). Furthermore, successful cochlear implantation for patients with Usher syndrome has been reported (Han et al, 2004; Loundon et al, 2003).




