Fundus Autofluorescence in Stargardt Disease
Vikki McBain
Noemi Lois
Stargardt disease (STGD) (1) (also termed fundus flavimaculatus [2]) is the most common recessively inherited macular dystrophy, affecting approximately one person in 10,000 (3). STGD can affect individuals of any gender and race (1,4, 5, 6, 7, 8) and there is wide variability in age of onset, visual acuity, fundus appearance, and severity of the disease (1,5, 6, 7, 8, 9, 10). Visual acuity may vary between 20/20 to 20/400; rarely will it drop below 20/400 (7,11). Patients with STGD may be asymptomatic or complain of visual acuity loss, photophobia, and, less commonly, nyctalopia (5).
Fundus examination may be normal in early stages of the disease or reveal retinal pigment epithelium (RPE) mottling or a bull’s-eye appearance at the macula, and active (deposition of yellow material at the level of the RPE) and/or resorbed (RPE depigmentation/atrophy) flecks and atrophy at the macula and midperipheral retina (1,5,6,9,12). Characteristically, the flecks have a pisciform (“fish-like”) appearance, but they can also be round, like dots, and appear either as individual lesions or joined together (6,13). Different clinical classifications of STGD have been proposed based on the presence or absence and distribution of the fundus lesions (6,9,12); however, none of these have been widely accepted.
Electrophysiology testing in patients with STGD may demonstrate macular dysfunction alone or macular and peripheral cone or cone and rod dysfunction (10,14). These patterns of functional loss cannot be predicted by the fundus appearance (10,14). Mutations in the ABCA4 gene, located in the short arm of chromosome 1, are responsible for all cases of STGD (15,16).
Currently, there is no treatment available for patients with STGD. However, laboratory studies suggest that progression of the disease may be slowed by protecting the eyes from light exposure (17). Additionally, new treatment strategies to reduce or prevent A2E accumulation in the RPE are also being investigated (see also Chapters 2 and 4) (Fig. 11G.1) (17, 18, 19, 20).
MOLECULAR BASIS AND PATHOLOGY
The mechanisms by which photoreceptors degenerate in STGD are not completely understood. Recent laboratory studies investigating the function of the ABCA4 protein, as well as studies conducted in the ABCA4 knockout mice, an animal model of the disease, have shed light on the molecular basis of STGD. The evidence suggests that the ABCA4 protein facilitates the transport of retinoids, preferentially N-retinylidene-phosphatidylethanolamine (N-retinylidene-PE) and all-trans-retinal (21,22), from the cytoplasmic side of the photoreceptor disc membrane to the cytosolic side, making them accessible to all-trans-retinol-dehydrogenase and facilitating its conversion into all-trans-retinol (22,23) (Fig. 11G.1A,B). This assists in the recovery of the photoreceptor cell following light exposure, reduces photoreceptor cell noise (the result of an increase in all-trans-retinal and opsin, which when combined can activate the visual transduction cascade), and diminishes the accumulation of all-trans-retinal and N-retinylidene-PE within the disc membranes (22). The latter in turn increases the production of N-retinylidene-N-retinyl-ethanolamine (A2E), the major fluorophore of lipofuscin, in the RPE (see also Chapter 2), which has potential cytotoxic effects on RPE cells (24, 25, 26, 27). RPE damage/loss is then followed by photoreceptor cell degeneration and loss of vision (33).
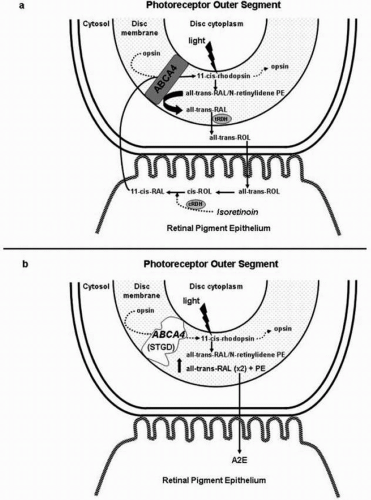 FIGURE 11G.1. (A) All-trans-retinal (all-trans-RAL) can react with phosphatidylethanolamine (PE) and form N-retinylidene-PE. Free all-trans-RAL and all-trans-RAL contained in N-retinylidene-PE are reduced to all-trans-retinol (all-trans-ROL) by the all-trans-retinol dehydrogenase (tRDH). Evidence suggests that ABCA4 transports N-retinylidene-PE and all-trans-RAL from the cytoplasmic side of the disc membrane to the cytosolic side, where they are reduced to all-trans-ROL by the all-trans-retinol DHase (trDH). (B) In patients with STGD, there is an impaired transport of all-trans-RAL and N-retinylidene-PE, with the subsequent accumulation of both molecules in the photoreceptor outer segment disc membrane. Condensation of all-trans-RAL and PE gives rise to N-retinylidene-N-retinyl-ethanola mine (A2E). A2E, the major fluorophore of lipofuscin, then accumulates in the RPE after photoreceptor outer segment disc shedding (modified from Refs. 28 and 57). CRDH, Cis-retinol DHase. |
Histopathological evaluation of eyes from patients with STGD have shown RPE cells densely packed with a substance with ultrastructural, autofluorescent, and histochemical characteristics consistent with lipofuscin in both the macula and peripheral retina (29,30). Only one histopathology study failed to detect increased lipofuscin in the RPE in a case of STGD without maculopathy (31). Subretinal desquamated RPE cells, macrophages engorged with melanolipofuscin in the outer retina, RPE and choriocapillaris atrophy, and photoreceptor-cell loss at the fovea have been also observed (29,30).
DIAGNOSTIC TECHNIQUES
Fundus flecks are the hallmark of STGD. Although “active” flecks are often seen by slit-lamp biomicroscopy or indirect ophthalmoscopy (5,9,32), “resorbed” flecks are more difficult to visualize and can be missed by the examining ophthalmologist. In the latter case, the diagnosis of STGD may be difficult.
Fluorescein and Indocyanine Green Angiography
Fluorescein angiography (FA) is only rarely required for the diagnosis or evaluation of patients with STGD. In both early and late frames of FA, “active” flecks appear hypofluorescent (5). “Resorbed” flecks may appear either hypo- (5) or hyperfluorescent (9). Areas of overt macular atrophy are visualized as areas in which no choriocapillaris is present but large choroidal vessels are seen. Patients with STGD may have a “dark choroid” or “choroidal silence” sign (33), characterized by a lack of early hyperfluorescence coming from the choroid, such that the retinal blood vessels, even the small capillaries, are easily seen over a very dark background where there is no choroidal fluorescence. Not all patients with STGD will demonstrate a dark choroid. In a recent study, only 62% of patients with STGD had this FA sign (7). Similarly, the dark choroid is not a specific sign of STGD; it has also been observed in patients with cone and cone-rod dystrophy (see also Chapter 11B) (7,33,34). However, if present, this sign may be useful in the differential diagnosis of STGD (see below), especially when the diagnosis of multifocal pattern dystrophy simulating STGD is entertained (see also Chapter 11E). Of interest, the overall increase in the fundus autofluorescence (AF) signal observed in patients with STGD (see below) seems to be independent of the presence or absence of a dark choroid (35).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


