Fundus Autofluorescence in Pattern Dystrophy
Susan M. Downes
Pattern dystrophy (PD), a term coined by Marmor and Byers (1), refers to a group of inherited retinal dystrophies characterized by the deposition of a highly autofluorescent material at the level of the retinal pigment epithelium (RPE), with an onset late in life. The incidence and prevalence are not known; however, PD is considered to be rare. In most cases there is an autosomal dominant (AD) mode of inheritance, although penetrance is variable. However, some cases may be sporadic or inherited as an autosomal recessive (AR) trait (the latter in association with reticular dystrophy). Patients may be asymptomatic or present with blurred vision and/or metamorphopsia. In advanced stages of the disease, reduced central vision and reading difficulties may be noted as a result of the development of atrophy or, less frequently, choroidal neovascularization (CNV).
The most common form of PD, adult vitelliform macular dystrophy (AVMD), is characterized by a bilateral solitary yellow, round to oval subfoveal lesion with or without a central pigmented spot (2). The next most common forms are multifocal PD simulating Stargardt disease-fundus flavimac ulatus (STGD-FFM) and butterfly-shaped PD (3,4). Rarer forms of PD include Sjogren reticular dystrophy, macroreticular dystrophy, and fundus pulverulentus. These various phenotypes can be seen in different individuals in the same family (5,6). Also, depending on the stage of the disease, the phenotype can change in appearance in the same individual over time (7,8).
PD may be present as an isolated condition or as part of a syndrome associated with other systemic disorders. PD has been found in a proportion of patients with maternally inherited diabetes and deafness (MIDDM) (9) (see Chapter 11H), myotonic dystrophy (10), pseudoxanthoma elasticum (PXE) (11), Friedreich ataxia (12), and Crohn disease (13).
HISTOPATHOLOGY AND MOLECULAR GENETICS
Clinicopathologic studies of patients with PD have shown similar findings (14, 15, 16). A histopathological study of the postmortem eyes of a 61-year-old female revealed focal atrophy of the RPE in the foveolar area bordered by hypertrophic RPE, with fusiform collagenous plaques of eosinophilic material located between the atrophic RPE and Bruch’s membrane. The sensory retina over the abnormal RPE displayed significant atrophy of the outer nuclear layer with loss of photoreceptor inner layer and outer segments. Pigment-laden macrophages with periodic acid Schiff-positive material had migrated into the atrophic outer retina. Ultraviolet fluorescent microscopy demonstrated massive accumulation of lipofuscin within the macular RPE as well as within the macrophages in the atrophic outer retina. Scanning electron microscopy revealed a confluent area of flattened atrophic RPE cells surrounded by
taller hypertrophic RPE cells. Transmission electron microscopy demonstrated that the RPE cells contained many lipofuscin granules (14,15). In three patients with AVMD, RPE and photoreceptor cell loss were observed in the central area. A moderate number of pigment-laden macrophages were present in the subretinal space and outer retina. The RPE was distended to both sides of the central lesion with abundant lipofuscin (16).
taller hypertrophic RPE cells. Transmission electron microscopy demonstrated that the RPE cells contained many lipofuscin granules (14,15). In three patients with AVMD, RPE and photoreceptor cell loss were observed in the central area. A moderate number of pigment-laden macrophages were present in the subretinal space and outer retina. The RPE was distended to both sides of the central lesion with abundant lipofuscin (16).
PD is genetically heterogeneous. To date, the most common genes known to harbor mutations that can cause nonsyndromic PD include PRPH2 (Peripherin 2) (Stone, personal communication referred to in Grover et al. [17]) (18), ELOVL4 (19), and BEST1 (20) (previously known as VMD2; see also Chapter 11F). In addition, as noted above, gene mutations in DTM (myotonic dystrophy) and mtDNA 3243 may be associated with a “PD-like” phenotype.
PRPH2 encodes peripherin 2, a membrane-associated glycoprotein restricted to photoreceptor outer segment discs (21). Normal levels of this protein are required for the morphogenesis and maintenance of the photoreceptor outer segments (22). The rds mouse exhibiting the retinal degeneration slow phenotype was found to have a single spontaneous mutation (23). The phenotype in the rds mouse is characterized by abnormal development of photoreceptor outer segments in the retina followed by a slow degeneration of the rods and cones, resembling the phenotypes described in human retinal dystrophies associated with PRPH2 mutations (24). A feature seen in both humans with PRPH2 mutations and mice with the rds mutation is a loss of photoreceptor function. Ali et al. (25) demonstrated that a subretinal injection of recombinant adeno-associated virus (rAAV) encoding an rds transgene resulted in the stable generation of outer segment structures and formation of new stacks of discs, which were morphologically similar to normal outer segments, and electrophysiological tests confirmed function. rAAV-mediated gene replacement of peripherin 2 restored retinal ultrastructure and function for as long as 14 weeks in the rds mouse. In this model, however, AAV-mediated gene replacement was not sustained in the long term.
Although studies have described a common consistent phenotype caused by certain mutations in peripherin 2, such as the Arg172Tryp mutation associated with macular dystrophy (26), significant variation in phenotypic expressivity in the same family has also been described in PD associated with PRPH2 mutations (5). For example, the same mutation can cause retinitis pigmentosa, PD, and fundus flavimaculatus in a single family (5). Yang et al. (4) described three separate families, each of which had a distinct PD phenotype that differed from those of the other families, although they all had the same mutation in PRPH2 and an identical disease haplotype.
ELOVL4 is a photoreceptor-specific gene that is involved in the biosynthesis of very long chain fatty acids. It is expressed in retina and skin. Defective protein trafficking may underlie the molecular mechanism associated with degeneration of the macula (27). It is possible that some patients with multifocal PD simulating STGDFFM have mutations in this gene, as mutations in ELOVL4 have been found in patients with AD Stargardt-like disease (19,28).
CLINICAL FEATURES
The age of onset in PD is usually in late adulthood, but with a positive family history it may occur in early adulthood. The progression of visual loss is usually slow, and most patients maintain reading vision until later in life. The condition can be quite
asymmetrical. Monitoring vision, especially in drivers, is important. Regular self-monitoring with an Amsler grid should be encouraged, and patients should be advised to seek ophthalmic review if sudden onset of blurring or distortion occurs, which could indicate the development of CNV. CNV was originally thought to be rare in PD (29), but is probably not uncommon (30,31). Macular atrophy has been observed both at presentation and over time in patients with PD (32).
asymmetrical. Monitoring vision, especially in drivers, is important. Regular self-monitoring with an Amsler grid should be encouraged, and patients should be advised to seek ophthalmic review if sudden onset of blurring or distortion occurs, which could indicate the development of CNV. CNV was originally thought to be rare in PD (29), but is probably not uncommon (30,31). Macular atrophy has been observed both at presentation and over time in patients with PD (32).
Adult Vitelliform Macular Dystrophy
AVMD is most commonly dominantly inherited, with incomplete penetrance and highly variable expression, but there are a significant number of sporadic cases. In one study, 91% of patients with AVMD had no family history (8). Furthermore, lesions may be unilateral in a high proportion of patients (8). Typical findings include a yellow (Fig. 11E.1) or pigmented central deposit, around which a depigmented halo is often seen (Fig. 11E.2). There may be a central spot alone or there may be a few deposits near the lesion or in the peripheral retina (Fig. 11E.3). The electro-oculogram (EOG) light rise is usually normal or mildly affected. Color vision may be abnormal.
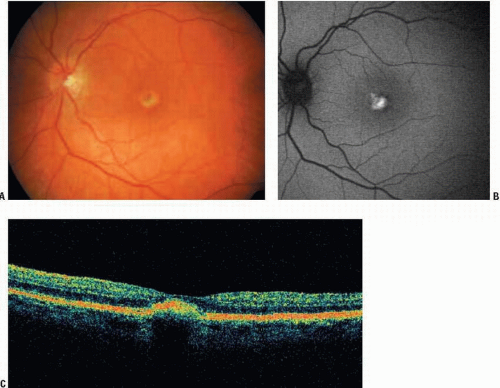 FIGURE 11E.1. Color fundus photograph (A), AF image (B), and OCT (C) obtained from a 64-year-old female with AVMD. Slit-lamp biomicroscopy disclosed a small yellow deposit at the left fovea (A), with a highly increased signal on AF imaging (B). OCT demonstrated a well-delineated, dome-shaped elevation of the anterior reflective band and increased reflectivity within the vitelliform lesion (C). PRPH2 testing demonstrated a mutation in the gene. |
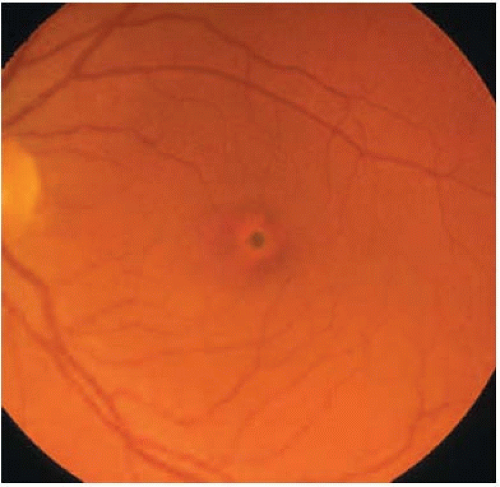 FIGURE 11E.2. Color fundus photograph obtained from a 56-year-old male with central visual disturbance and AVMD. A central area of increased pigmentation surrounded by a halo of depigmentation is observed. |
Butterfly-Shaped Pattern Dystrophy
Butterfly-shaped PD is characterized by an accumulation of yellow or brown pigment at the level of the RPE in a butterfly configuration (Fig. 11E.4). A subnormal EOG and normal or slightly diminished visual acuity have been described, as well as atrophic changes and an AD inheritance (3). Although Deutman proposed a relatively benign course for butterfly-shaped PD, it has now clear that butterfly-shaped PD, like other types of PD, is usually a progressive disorder with varying degrees of visual deterioration (3). Older individuals may have atrophic, depigmented lesions extending into the peripapillary region, with markedly reduced visual acuity (33).
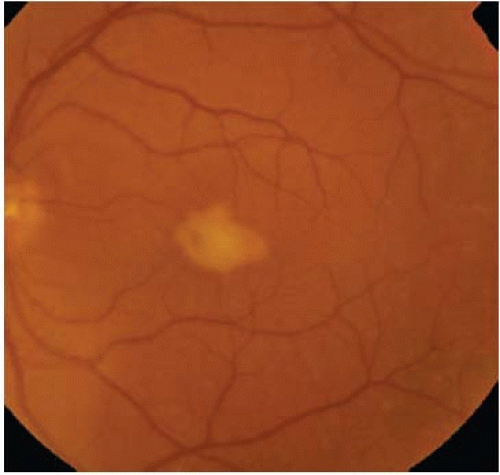 FIGURE 11E.3. Color fundus photograph of a patient with AVMD demonstrating accumulation of yellow material at the fovea and additional deposits near this main lesion distributed throughout the macula and into the midperipheral retina.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|