Fundus Autofluorescence in Maternal Inherited Diabetes and Deafness
Caren Bellmann
Pamela Rath
Mitochondria are ubiquitous in eukaryotes and are essential for survival. Their primary function is to support aerobic respiration and provide energy to the cell. Mitochondria also play an important role in cell signaling for apoptotic cell death. Genes located in the mitochondrial DNA (mtDNA) encode subunits of the mitochondrial respiratory chain where ATP is generated. Because pathogenic mutations of mtDNA usually do not affect all mtDNA within a cell, the clinical phenotype of the mitochondrial disease depends on the relative proportion of mutant and wild-type mtDNA in different tissues. Furthermore, depending on the energy demand of a particular cell, the level of mutated genomes required to produce a phenotypic expression will vary. Disease is expressed when, at a particular threshold, ATP production falls below the energy demand. Until relatively recently, there was a general lack of awareness of mitochondrial disease. Maternal inherited diabetes and deafness (MIDD) was first reported only in 1992 (1,2).
The prevalence of MIDD described in the general population is 0.06%; the disease is found in about 1.5% of diabetic populations in different countries and people of different ethnic backgrounds (1, 2, 3, 4). MIDD is considered to be a subtype of diabetes mellitus that cosegregates with the most common mitochondrial DNA point mutation, an adenine-to-guanine transition at position 3243 of the mitochondrial DNA (A3243G) (1).
Clinically, mitochondrial diabetes is typically combined with neurosensory hearing loss and retinal dystrophy. Therefore, this disease is easily distinguishable from the idiopathic forms of diabetes (4). Additional findings, such as a progressive defect in insulin secretion and neuromuscular signs, may be helpful in identifying the disease. The final diagnosis is made by detecting the mitochondrial DNA point mutation A3243G in peripheral blood leukocytes by molecular-genetic procedures using DNA from oral mucosa cells, hair follicles, or muscle biopsy.
Diabetic retinopathy changes are seldom found in patients with MIDD, whereas retinal dystrophy changes are very common. The latter have been described with a prevalence of up to 85.7% (5,6), and they may lead to a diagnosis of the condition. These retinal changes are concentrated on the posterior pole and range from mildly abnormal pigmentation at the posterior pole to extensive atrophy of the retinal pigment epithelium (RPE) with a symmetric distribution between both eyes (Fig. 11H.1) (5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19). Different patterns of pigmentary retinopathy have been observed within the same family, which suggests that the particular pattern present in a patient may change over time (11). Furthermore, in a recent study of seven patients with MIDD, 10 out of 34 maternal relatives presented the typical retinal changes of MIDD with abnormal pigmentation at the posterior pole. All of them tested positive for the A3243G mutation, suggesting that retinal abnormalities may be a reliable clinical indicator for a positive mutation (20). Despite the retinal changes, visual prognosis is generally good in patients with MIDD. In a multicenter study, 80% of patients presented with visual acuity of 6/7.5 or better in
both eyes (5). No correlation has been found among the severity of retinal changes, the patient’s age, and the percentage of mutant mtDNA (11). Moreover, there is no known correlation between the proportion of mutant mtDNA and the clinical features of family members (14). However, the percentage of mutant mtDNA may vary from one tissue to another and may not correspond to the percentage of mutant mtDNA in retinal tissue in a patient with this disease.
both eyes (5). No correlation has been found among the severity of retinal changes, the patient’s age, and the percentage of mutant mtDNA (11). Moreover, there is no known correlation between the proportion of mutant mtDNA and the clinical features of family members (14). However, the percentage of mutant mtDNA may vary from one tissue to another and may not correspond to the percentage of mutant mtDNA in retinal tissue in a patient with this disease.
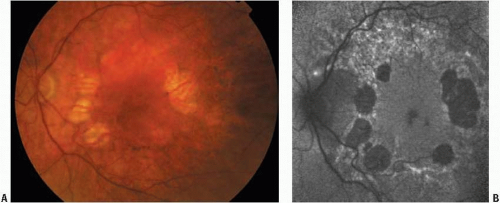 FIGURE 11H.1. Fundus photograph (A) and AF image (B) obtained from the left eye of a patient with MIDD. Patches of RPE atrophy preserving the fovea were observed on fundus examination (A). AF images disclosed well-defined areas of reduced AF signal (B). AF images defined more clearly areas of RPE loss. In fact, at a location where clinically there seemed to be RPE atrophy (superior to the fovea), AF images disclosed a background AF signal, indicating preserved AF and thus RPE in this area. Very small foci of increased and decreased AF signal are also visible outside the fovea and in between areas of atrophy. |
MOLECULAR BASIS AND PATHOLOGY
The pathogenesis of the retinal dystrophy in MIDD is not clear. Retinal pigmentary abnormalities commonly occur in other mitochondrial disorders, such as Kearns-Sayre syndrome, a neuromuscular disorder that is characterized by, in addition to retinal changes, chronic progressive external ophthalmoplegia, and heart disease. In MELAS syndrome, the combination of mitochondrial myopathy, encephalomyopathy, lactate acidosis, and stroke-like episodes leads to a high prevalence of retinal changes (21). Both syndromes are caused by the same mitochondrial A3243G mutation as described for MIDD. Morphological studies in patients with Kearns-Sayre and MELAS syndromes revealed ultrastructural changes in the RPE with enlarged mitochondria (22). Pathologic abnormalities were most marked posteriorly and included both hypo- and hyperpigmentation (23, 24, 25, 26). These studies showed further degeneration of photoreceptor outer segments as well as complete atrophy of photoreceptor cells. It was suggested that these changes were most likely secondary to RPE degeneration. Chang and coworkers (24) reported ocular histopathologic and ultrastructural changes in two patients who suffered from Kearns-Sayre and MELAS syndromes. In one of the patients studied, fluorescein angiography (FA) had been obtained 3 years before the patient’s death. The blocked fluorescence described on FA was thought to be due to the accumulation of lipofuscin in RPE cells. Pathologic evaluation of the same eye revealed a degenerated RPE with overlying photoreceptor cell atrophy in the central retina. In adjacent areas, there was intact RPE containing melanin granules, lipofuscin globules, and large melanolipofuscin conglomerates
with overlying disorganized photoreceptor outer segments. However, McKechnie and coworkers (23) could not confirm an increased amount of lipofuscin in RPE cells in a case with Kearns-Sayre syndrome.
with overlying disorganized photoreceptor outer segments. However, McKechnie and coworkers (23) could not confirm an increased amount of lipofuscin in RPE cells in a case with Kearns-Sayre syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


