The endolymphatic sac tumor is a slow-growing, locally aggressive neoplasm that originates from the epithelium of the endolymphatic sac and duct. Disease progression can lead to profound sensorineural hearing loss, posterior fossa invasion, brainstem compression, drop metastasis, and eventual death. Early diagnosis and surgical attention are the primary objectives in the management of patients who have endolymphatic sac tumor. This article describes the latest rationale and techniques for hearing preservation surgery and a review of the latest developments surrounding this disease entity.
Endolymphatic sac tumor (ELST) is a slow-growing, locally aggressive neoplasm that originates from the epithelium of the endolymphatic sac and duct. It demonstrates the histologic appearance of a papillary adenomatous lesion, and before its acceptance as a disease entity, its destructive behavior within the temporal bone was attributed in some cases to a metastatic renal cell carcinoma variant . By virtue of ELST-related obstruction of the vestibular aqueduct, patients often develop symptoms of endolymphatic hydrops , and this pathologic finding recently was confirmed histologically . The earliest manifestations of ELSTs are often hearing loss and vertigo that initially may lead to a misdiagnosis of Meniere’s disease. Disease progression can lead to profound sensorineural hearing loss, posterior fossa invasion, brainstem compression, drop metastasis, and eventual death. Although ELSTs are known to occur more frequently in patients with von Hippel-Lindau disease (VHL), a diagnosis of VHL is not a prerequisite, because these tumors also appear sporadically in patients who do not have VHL. Early diagnosis and surgical attention are the primary objectives in the management of patients who have ELST because recent studies have shown the potential for hearing preservation when small tumors are removed successfully.
Historical background
ELST is a recently recognized neurotologic disease entity characterized by the presence of a destructive papillary cystic adenomatous tumor of the temporal bone. Its roots and evidence for its existence date to early in the last century in a report that described a destructive lesion of the temporal bone in a patient who had VHL that was ascribed to metastatic renal cell carcinoma in the absence of a primary kidney tumor . Throughout the ensuing years it became clear that an aggressive papillary tumor could arise primarily within the temporal bone; however, the site of epithelial origin remained an area of conjecture. This uncertainty was partly caused by the fact that many of these cases occurred in patients with an intact tympanic membrane and normal external auditory canal, so the glandular epithelial tissue of the external auditory canal was excluded as the site of origin.
In 1989, Heffner studied 20 such large papillary cystic tumors of the temporal bone and concluded that the endolymphatic sac epithelium represented the most likely area at which these lesions initially grow. This finding was corroborated by an earlier study in 1984 by Hassard and colleagues , who described a papillary cystic lesion filling the endolymphatic sac that was encountered during decompression surgery for presumed Meniere’s disease and likely represented the first clinical confirmation of the validity of the ELST designation. In 1993, Li and colleagues formally proposed that the designation for aggressive papillary tumors of the temporal bone be changed to ELST.
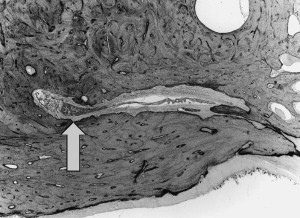
In 1995, Megerian and colleagues provided temporal bone histologic evidence of a de novo ELST tumor in a patient who had VHL ( Fig. 1 ) and died of complications related to a massive ELST of the contralateral ear. Included in that study were seven additional clinical cases that revealed that most of the patients had Meniere’s-like symptoms years before the diagnosis of ELST, which raised the possibility that early manifestations of tumor growth included perturbations of vestibular aqueduct function and the development of endolymphatic hydrops-related symptomatology. Poe and colleagues and later Manski and colleagues studied ELST in patients who had VHL, and the latter group documented the fact that at least 13% of patients who had VHL harbored ELST tumors. They suggested that patients who had VHL should undergo regular surveillance, because undetected growth was shown to result in deafness that sometimes was bilateral.
By 1997, screening for ELST using MRI became standard for patients who had VHL, whereas before that time annual MRI evaluations of the head focused mainly on detection of VHL-related cerebellar hemangioblastomas. These practices, coupled with an increased number of reports in the literature of sporadic ELSTs, led to the evolution of surgical alternatives for tumor removal. In 2002, Megerian and colleagues described techniques for tumor removal in small ELSTs that afforded hearing preservation. Subsequent reports by Hansen and Luxford in 2004 and Kim and colleagues in 2005 confirmed the clear role for hearing preservation surgery using the retrolabyrinthine approach for small tumors and more aggressive skull base extirpations for larger lesions.
Etiology
As recently as 2004, Bambakidis and colleagues reported that most ELSTs described in the literature have occurred sporadically in patients who do not have VHL compared with patients who do (103 versus 46). Much information that has been learned recently regarding the genesis of these lesions has been via an understanding of the genetics and molecular biology that result from a mutation in the VHL tumor suppressor gene. VHL is an autosomal dominant multisystemic disorder characterized by cerebellar hemangioblastomas, retinal angiomas, and renal cysts, clear cell renal carcinomas, and other visceral tumors.
In the late 1990s, ELST became a recognized part of the disease spectrum because it was associated with online Mendelian inheritance in man VHL disease (No. 193300) . VHL has a prevalence of 1 in 39,000 people and is caused by a germline mutation in the VHL gene on chromosome 3 (3p25-26) . The VHL gene product is a tumor suppressor that when lost or mutated results in a loss of inhibition to cell growth and can lead to neoplasms such as ELST. The VHL gene product protein is important in the regulation of hypoxia inducible factor–1α, which controls angiogenesis and cell metabolism .
In normal situations, patients have two copies of the wild-type (normal) VHL gene allele. The disease develops in people who have VHL and have a spontaneous “loss of heterozygosity,” that is, a mutation of the normal copy of the VHL gene, which leaves only the mutated copy of the VHL gene . This mutation leads to tumorigenesis and the development of the highly vascular ELST in vulnerable tissue, such as the epithelium of the vestibular aqueduct. Recent studies of clinically tumor-free endolymphatic duct and sac tissue of patients who have VHL revealed VHL gene–deficient microscopic abnormalities with morphologic similarities to ELST not only within the sac epithelium but also within the duct . The implications of this finding are important, because it confirms prior histologic observations regarding a de novo ELST reported in 1995, which showed a tumor filling not only the proximal sac but also the intraosseous duct, and it underscores the need for surgery to include removal of the duct for long-term cure .
The propensity for microscopic tumor foci along the duct also may explain the observation that as many as 65% of patients who have VHL demonstrate an audiologic disturbance despite only 6% of those patients showing MRI evidence of a tumor . It is likely that these tumors remain radiologically undetectable for some time, causing hearing and balance disturbance with an average delay of 15 years before tumor is detected . It follows that patients who do not have VHL who develop ELST likely have suffered a “two-hit” injury to the VHL locus and have lost function of both copies of the wild-type VHL tumor suppressor allele.
Pathophysiology
Whether an ELST develops in a patient who has VHL or sporadically in a patient without it, the pathologic sequence of growth seems to advance slowly, over the course of years. Initially, a small tumor within the confines of the endolymphatic sac and duct (usually the distal duct and proximal sac) causes audiovestibular dysfunction that can take one of two courses. One such course is an acute significant hearing loss, which may be irreversible and has been shown to likely be secondary to intralabyrinthine hemorrhage followed by inflammation and neural degeneration . More commonly, symptoms of endolymphatic hydrops develop, which result in low frequency hearing loss, tinnitus, fullness, and vertigo . This development is likely caused by blockage of endolymphatic fluid resorption or even excess fluid production that results in classic endolymphatic hydrops and frequent early diagnosis of Meniere’s disease . As the tumor grows, it takes on the appearance of an infiltrative, poorly circumscribed neoplasm composed of cuboidal to low-columnar epithelial cells disposed in a fibrous stroma and arranged either in papillary patterns ( Fig. 2 ) or cystic spaces that contain proteinaceous material reminiscent of thyroid follicles . The epithelial cells are often clear and are sometimes arranged in follicular configurations; however, thyroglobulin stains are negative .
By this time, an enlarging tumor typically has eroded the osseous vestibular aqueduct and proceeds to grow along four potential vectors of extension from the endolymphatic sac . In order of frequency, ELSTs progress posteriorly to the cerebellopontine angle through the posterior fossa dura, laterally via the mastoid air cell tracts to the middle and external ear (often with facial paralysis), superiorly toward and into the middle cranial fossa (usually with destruction of semicircular canals) ( Fig. 3 ), and finally along the petrous ridge to the clivus and cavernous and sphenoid sinuses. Before recognition of the ELST entity, the finding of these lesions in the temporal bone and cerebellopontine angle was thought by some to be the result of ectopic choroid plexus papillomas because of the similar appearance of aggressive papillary tumors (later believed to be ELSTs) of the temporal bone and tumors of the choroids plexus. The ELSTs were shown to lack reliable immunohistochemical staining for transthyretin, however, which is an important marker for choroid plexus and is readily expressed in choroid plexus papillomas . Nearly all ELSTs express cytokeratin, vimentin, and epithelial membrane antigen, and most stain positive for S-100 and neuron-specific enolase .
Etiology
As recently as 2004, Bambakidis and colleagues reported that most ELSTs described in the literature have occurred sporadically in patients who do not have VHL compared with patients who do (103 versus 46). Much information that has been learned recently regarding the genesis of these lesions has been via an understanding of the genetics and molecular biology that result from a mutation in the VHL tumor suppressor gene. VHL is an autosomal dominant multisystemic disorder characterized by cerebellar hemangioblastomas, retinal angiomas, and renal cysts, clear cell renal carcinomas, and other visceral tumors.
In the late 1990s, ELST became a recognized part of the disease spectrum because it was associated with online Mendelian inheritance in man VHL disease (No. 193300) . VHL has a prevalence of 1 in 39,000 people and is caused by a germline mutation in the VHL gene on chromosome 3 (3p25-26) . The VHL gene product is a tumor suppressor that when lost or mutated results in a loss of inhibition to cell growth and can lead to neoplasms such as ELST. The VHL gene product protein is important in the regulation of hypoxia inducible factor–1α, which controls angiogenesis and cell metabolism .
In normal situations, patients have two copies of the wild-type (normal) VHL gene allele. The disease develops in people who have VHL and have a spontaneous “loss of heterozygosity,” that is, a mutation of the normal copy of the VHL gene, which leaves only the mutated copy of the VHL gene . This mutation leads to tumorigenesis and the development of the highly vascular ELST in vulnerable tissue, such as the epithelium of the vestibular aqueduct. Recent studies of clinically tumor-free endolymphatic duct and sac tissue of patients who have VHL revealed VHL gene–deficient microscopic abnormalities with morphologic similarities to ELST not only within the sac epithelium but also within the duct . The implications of this finding are important, because it confirms prior histologic observations regarding a de novo ELST reported in 1995, which showed a tumor filling not only the proximal sac but also the intraosseous duct, and it underscores the need for surgery to include removal of the duct for long-term cure .
The propensity for microscopic tumor foci along the duct also may explain the observation that as many as 65% of patients who have VHL demonstrate an audiologic disturbance despite only 6% of those patients showing MRI evidence of a tumor . It is likely that these tumors remain radiologically undetectable for some time, causing hearing and balance disturbance with an average delay of 15 years before tumor is detected . It follows that patients who do not have VHL who develop ELST likely have suffered a “two-hit” injury to the VHL locus and have lost function of both copies of the wild-type VHL tumor suppressor allele.
Pathophysiology
Whether an ELST develops in a patient who has VHL or sporadically in a patient without it, the pathologic sequence of growth seems to advance slowly, over the course of years. Initially, a small tumor within the confines of the endolymphatic sac and duct (usually the distal duct and proximal sac) causes audiovestibular dysfunction that can take one of two courses. One such course is an acute significant hearing loss, which may be irreversible and has been shown to likely be secondary to intralabyrinthine hemorrhage followed by inflammation and neural degeneration . More commonly, symptoms of endolymphatic hydrops develop, which result in low frequency hearing loss, tinnitus, fullness, and vertigo . This development is likely caused by blockage of endolymphatic fluid resorption or even excess fluid production that results in classic endolymphatic hydrops and frequent early diagnosis of Meniere’s disease . As the tumor grows, it takes on the appearance of an infiltrative, poorly circumscribed neoplasm composed of cuboidal to low-columnar epithelial cells disposed in a fibrous stroma and arranged either in papillary patterns ( Fig. 2 ) or cystic spaces that contain proteinaceous material reminiscent of thyroid follicles . The epithelial cells are often clear and are sometimes arranged in follicular configurations; however, thyroglobulin stains are negative .



