ascending helix; some believe that these structures can arise from the second arch (3). By approximately 18 weeks’ gestation, the auricle has achieved essentially adult form, although it continues to grow in childhood with changes continuing into late adult life.
TABLE 140.1 OVERVIEW OF EAR DEVELOPMENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
with variable expression and incomplete penetrance have been reported. More recently, specific genes, such as the
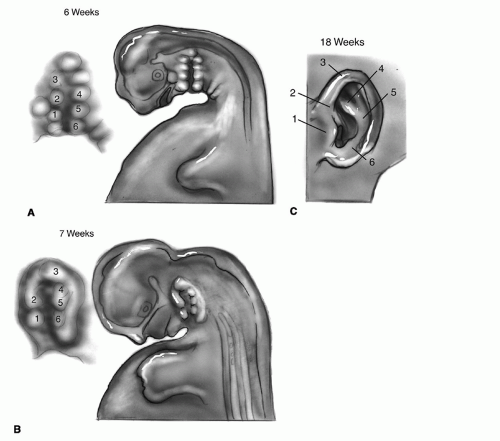 Figure 140.1 Development of the auricle. A: Six hillocks form on the first and second branchial arches. All can be identified at 6 weeks’ gestation. B: Seven-week stage. C: By 18 weeks, the adult form is recognizable. |
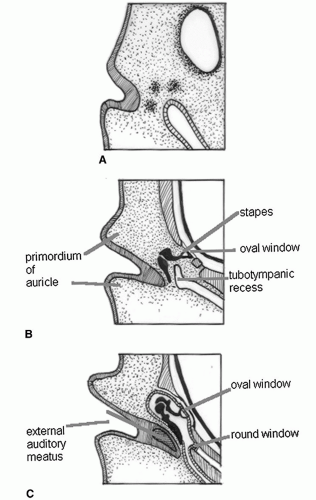 Figure 140.2 Development of the middle ear and ear canal. A: Week 5. B: Week 10. C: Week 27. |
ear. Associated auricular abnormality exists in 94% of cases of atresia, and the middle ear is frequently deranged (11), in part because all these structures are derived from the first two branchial arches and the intervening branchial cleft. Consequent are further abnormalities of branchial arch derivatives, such as the mandible. Among children with microtia or anotia, 20% to 40% have an identifiable syndromal malformation, such as hemifacial microsomia, Treacher Collins syndrome (mandibulofacial dysostosis), or Goldenhar (oculoauriculovertebral) syndrome (5, 12). These syndromal malformations are discussed in Chapter 86.
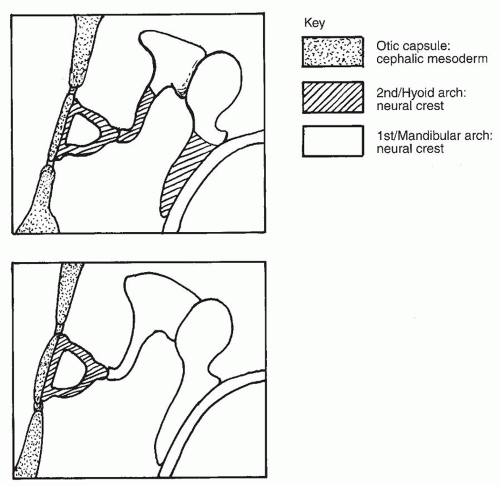 Figure 140.3 Origin of the ossicles—two interpretations. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


