Congenital Malformation of the Ear
Paul R. Lambert
Atresia of the ear canal with middle ear anomalies can occur in isolation or in association with microtia or craniofacial dysplasia. The reported incidence is 1 in 10,000 to 20,000 births. Genetic transmission occurs in many of the syndromes that include aural atresia (e.g., Treacher Collins syndrome), but it is rarely found in cases of isolated atresia. Aural atresia is bilateral in approximately one-third of the cases, and each side can vary in complexity (1).
The evaluation and treatment of aural atresia present a number of challenges to the otologic surgeon. First, overall hearing must be assessed and the need for immediate amplification determined. The second challenge is to formulate a long-term rehabilitation strategy. The key component of this challenge is to determine if surgical correction of the atresia is appropriate. This decision process requires the integration of results from audiometric and radiographic studies with a qualitative assessment of the patient’s functional hearing status and the probability of restoring serviceable hearing. If surgery is recommended, the last challenge becomes the operative procedure itself, which is made complex by abnormal development of the temporal bone. This fact places the facial nerve and labyrinth at a greater risk than that encountered in routine temporal bone surgery and complicates the healing process, particularly for canal patency. This chapter reviews the concepts and protocols necessary to meet these challenges successfully.
EMBRYOLOGY
Placed in the context of congenital aural atresia, a general knowledge of the embryologic development of the ear is fascinating and essential for understanding the altered surgical anatomy. Development of the first pharyngeal pouch, the first and second branchial arches, the first branchial cleft, and the otic capsule must all be considered in this discussion.
External Auditory Canal
The external ear canal is derived from the first branchial groove (cleft), between the mandibular and hyoid arches. It is initially represented by a solid core of epithelial cells that extends down to the area of the tympanic ring and first pharyngeal pouch. This core of cells remains in place until the middle trimester of fetal life, a time when most structures of the inner, middle, and outer ear are well differentiated. At this point, absorption of the epithelial cells begins, progressing in a medial to lateral direction. If this canalization process is arrested prematurely, it is possible to have a more normally developed tympanic membrane and bony external ear canal associated with an atretic or very stenotic membranous canal, a situation that predisposes to canal cholesteatoma formation as the trapped squamous epithelium continues to desquamate.
The medial portion of the external ear canal is formed by the tympanic bone. This structure begins to ossify in the third embryonic month, eventually forming the tympanic ring and osseous ear canal; the latter structure continues its lateral growth during the first and second postnatal years. Malformation of the tympanic bone produces atretic bone at the level of the tympanic membrane and results in atresia of the ear canal (2). The mandibular condyle articulates with this rudimentary tympanic bone.
Mastoid and Middle Ear
The eustachian tube, middle ear, and mastoid air cells are derived from the first pharyngeal pouch. Although the middle ear cavity and mastoid air cells are smaller than normal in patients with aural atresia, no anatomic or clinical studies show impaired eustachian tube function in these ears. Pneumatization of the mastoid is a late embryologic event, starting at the seventh or eighth month and continuing into postnatal life. A well-pneumatized mastoid usually
indicates good middle ear development, including size of the tympanum and formation of the ossicles. The relationship between middle ear development and degree of differentiation of the pinna is disputed (3).
indicates good middle ear development, including size of the tympanum and formation of the ossicles. The relationship between middle ear development and degree of differentiation of the pinna is disputed (3).
The ossicles, except for the vestibular portion of the stapes footplate, are formed from the first and second branchial arches. The external ear canal and tympanic membrane are derived from the first branchial cleft. Isolated branchial arch (ossicular) or branchial cleft (external ear canal) deformities are possible, but usually these malformations occur in combination (2). Other branchial arch defects may occur, especially mandibular hypoplasia. The stapes footplate is formed in part from the otic capsule, and in most cases it is normally developed in ears with congenital atresia. It is uncommon to encounter a fixed stapes footplate in the usual major congenital ear malformation, although the superstructure is frequently deformed. This information is important, because the stapes is often partially obscured by the lateral ossicular mass or the facial nerve in atretic ears, and its normal mobility may be difficult to determine with certainty.
Absence of the stapes footplate and oval window can occur, but this is usually encountered in a patient with a patent external ear canal and normal-appearing tympanic membrane rather than in a patient with aural atresia (4). It has been suggested that this condition is caused by abnormal development of the facial nerve (5, 6). By the fifth to sixth week of gestation, the horizontal and vertical segments of the facial nerve are evident (6). If anterior displacement of the facial nerve occurred at this time, the nerve could become interposed between the otic capsule and the stapes blastema, which is beginning to grow toward the otic capsule. This would interfere with further stapes development, resulting in a rudimentary ossicle attached to the incus. Continued growth of the stapes toward the otic capsule could result in the rudimentary crura becoming embedded in the displaced facial nerve (4). Because the stapes never contacts the otic capsule, an oval window does not form. With further anterior displacement of the facial nerve, it is possible for that structure to course across the promontory, inferior to the region of the oval window (4). It has been hypothesized that displacement of the facial nerve occurs because of underdevelopment of the first branchial arch (5). This results in a compensatory overshifting of the second branchial arch, and its nerve follows this shift, assuming a more anterior position.
Inner Ear
The membranous labyrinth is derived from the ectodermal otocyst. In most cases of isolated aural atresia, sensorineural and vestibular functions are normal. A comprehensive review of inner ear abnormalities in aural atresia was recently reported by Vrabec and Lin (7). Their study included aural atresia within a broad spectrum of conditions termed “craniofacial microsomia” (e.g., CHARGE, branchiootorenal syndrome, Treacher Collins syndrome). They noted an inner ear anomaly by computed tomography (CT) in 22% of 105 patients, with an abnormality of the horizontal semicircular canal and/or vestibule accounting for over half (Fig. 148.1). A sensorineural hearing loss, usually mild and defined as a bone-conduction threshold greater than 20 dB involving two or more frequencies, was found in 15% of patients; the sensorineural hearing loss was usually mild.
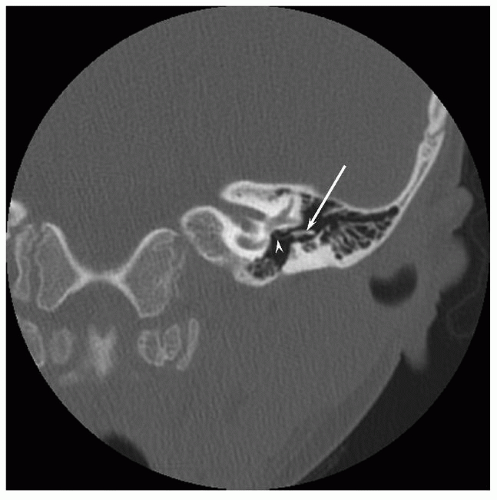 Figure 148.1 Aural atresia with inner ear dysplasia. Axial CT shows absence of the external auditory canal, incomplete partition of the upper turns of the cochlea (arrow), and an enlarged lateral semicircular canal (arrowhead). |
Facial Nerve
Preoperative facial nerve weakness is uncommon in isolated aural atresia, but can be seen in 10% to 15% of patients with aural atresia in association with other craniofacial anomalies (7). Facial nerve abnormalities are common in cases of aural atresia (8, 9). Bony dehiscence of the fallopian canal frequently occurs, and the facial nerve may also take an anomalous course. Typically, the facial nerve makes an acute angle at the second genu, crossing the middle ear in a more anterior and lateral direction to exit into the glenoid fossa. This abnormal position of the mastoid segment of the facial nerve places it at jeopardy when drilling the posterior inferior portion of the new external ear canal. As the facial nerve exits the skull, it may lie just deep to the area of the tragus. Inadvertent injury to the nerve can occur if undermining of the auricle is necessary to better align the meatus and newly created external ear canal. A correlation between the degree of microtia and the extent of facial nerve abnormality has been observed (3, 10). An association between preoperative facial nerve dysfunction and inner ear abnormality by CT has also been noted (7).
CLASSIFICATION
Patients with congenital aural atresia are classified on the basis of auricular development and external canal/middle ear development. Deformity of the auricle is straightforward and is divided into three grades (11). Grade I microtia represents a minor malformation, with the auricle being smaller than normal but with all parts discernible. In grade II microtia, the auricle is represented by a curving or vertical ridge of tissue. In grade III microtia, any resemblance to an auricle is lost, and only a small rudimentary soft tissue structure is present.
Classification of the external canal/middle ear deformity has been more problematic because of the various parameters that have been used, including clinical examination, radiographic findings, surgical observations, or histopathologic studies. Ombredanne (12) proposed dividing congenital aural atresia into two groups only, major and minor malformations. This classification scheme is attractive because of its simplicity and clinical utility. With minor modifications, the following descriptions reflect Ombredanne’s criteria.
Major Malformation
In the major malformation group, the external ear canal and tympanic membrane are usually absent, although cases of severe canal stenosis are also included. A small rudimentary tympanic membrane attached to a bony septum is occasionally seen in the canal stenosis patients, but typically the stenosis prevents visualization of the medial aspect of the ear canal. The size of the middle ear space is reduced, and the malleus and incus are deformed, fused, and fixed to the atretic bone. In severe cases, the middle ear space is very hypoplastic, and ossicles are rudimentary or absent. Dehiscence or displacement of the facial nerve can be expected in most major malformations. Grade II or III microtia is common, and inner ear function is usually normal.
Minor Malformation
The significant defect in the minor malformation group involves the middle ear. A conductive hearing loss exists because of absence or deformity of one or more ossicles or fixation of the ossicular chain. Abnormalities of the stapes may be more severe in the minor malformation group than in the major group. The middle ear space and tympanic membrane are normal or only slightly smaller. The external ear canal is patent but may be mildly stenotic. Dehiscence or displacement of the facial nerve can occur, and the pinna is normally developed or only slightly deformed.
PATIENT EVALUATION
Most cases of major congenital ear malformations are evident at birth because of microtia or other craniofacial anomalies. Patients with a normal or only slightly deformed pinna and a stenotic or blindly ending external auditory canal, however, may escape diagnosis for years. Unilateral minor congenital ear malformations with a patent ear canal and normal tympanic membrane can be more difficult to diagnose and may be discovered only with routine hearing screening in school.
The evaluation of an infant or young child with congenital aural atresia must involve both the functional and cosmetic aspects of this condition. First, one must assess the overall hearing status and need for immediate amplification. Second, one must formulate a treatment plan that usually includes consultation with other specialists (e.g., plastic surgery, genetics, developmental pediatrics.)
PHYSICAL EXAMINATION
The initial focus of the physical examination is on overall craniofacial development, because abnormalities or syndromes involving the first or second branchial arch may be associated with the aural atresia. Careful palpation of the mandible may reveal a mild hemifacial microsomia not immediately obvious on inspection. Development of the palate and other intraoral structures should be assessed. The degree of microtia is observed, as is mastoid development. The caliber of the external auditory canal should be graded as normal, stenotic (i.e., mild or severe), blindly ending, or completely atretic. If possible, the tympanic membrane is examined otoscopically. Displacement of the malleus handle (usually anteriorly) or a bony shelf extending from the posterior canal wall may exist. Mobility of the tympanic membrane and malleus handle is determined. Any abnormalities of the digits, limbs, or cervical vertebra are noted.
In the case of a child, achievement of neurologic milestones, such as speech and ambulation, is assessed by history and direct observation. This information can provide insight into auditory and vestibular development. Each major division of the facial nerve is carefully examined and any weakness observed. It is rare to encounter a paresis or paralysis involving the entire hemiface, although there is occasionally involvement of the lower face or lip area. The most common anomaly of facial function is a congenital absence of the depressor anguli oris muscle.
Audiometric Evaluation
Auditory assessment in patients with unilateral atresia is usually straightforward. Behavioral audiometry can be used in most cases, although auditory brainstem response testing may be necessary in young infants or children who are difficult to test. Patients with bilateral atresia present more of a challenge because of the masking dilemma. In such cases, it is essential to determine the level of cochlear function in each ear to prevent operating on an only-hearing ear or on an ear with little or no potential for hearing improvement. It is unsafe to assume that cochlear
function is normal bilaterally, even if inner ear development appears normal by CT. Objective data are needed, and bone-conduction auditory brainstem response testing can provide this (13). This testing involves placing recording electrodes near both ears to measure the responses to a bone-conduction stimulus.
function is normal bilaterally, even if inner ear development appears normal by CT. Objective data are needed, and bone-conduction auditory brainstem response testing can provide this (13). This testing involves placing recording electrodes near both ears to measure the responses to a bone-conduction stimulus.
Wave I of the auditory brainstem response is generated by the distal portion of the auditory nerve. There is minimal crossover of this small potential to the contralateral ear, and it is thus best measured by a recording electrode ipsilateral to the stimulated side. When recording simultaneously from both ears with surface electrodes, the presence of a wave I should represent the response from the ear being stimulated only. Although stimulation of each ear independently is not possible with a bone-conducted signal, the wave I response is ear specific, thereby allowing differential assessment of cochlear function.
If the ear canals are patent, electrocochleography can be used in a similar way to obtain ear-specific information. Instead of surface electrodes, a transtympanic, tympanic membrane, or canal electrode are possible, providing a more robust wave I response.
Computed Tomography
CT of the temporal bone is necessary in all patients being considered for surgery. It is also recommended in patients with stenosis of the external auditory canal to examine for possible cholesteatoma formation. To completely assess middle ear development, images in the axial (i.e., parallel to the line from the infraorbital rim to the external meatus) and coronal (i.e., parallel to the ramus of the mandible) planes are necessary. For example, the body of the malleus and incus, the incudostapedial joint, and the round window are best seen on axial images, but the stapes, the oval window, and the vestibule are best delineated on coronal images; both planes are necessary to follow the course of the facial nerve. Modern CT scanners can provide images in both planes without having to irradiate the patient twice. A 64-slice CT scanner (or better) is preferred to create diagnostic reformatted coronal images.
Assuming normal sensorineural function has been confirmed audiometrically, the decision to operate depends primarily on the degree of middle ear development, as reflected by the size of the tympanum and status of the ossicles. CT is also important for assessing the development of the cochlear and vestibular labyrinths because their appearance can influence middle ear surgery. For example, an enlarged vestibule and horizontal semicircular canal suggest the possibility of an abnormal communication between the perilymph and cerebrospinal fluid. In such cases, manipulation of the stapes should be avoided.
The course of the facial nerve usually can be delineated by CT. The inability to define this structure precisely, however, is not a contraindication to surgery, assuming that the other criteria for middle ear development are met.
Cholesteatoma can occur in association with congenital aural atresia. Occasionally, sufficient canalization of the external canal occurs such that a space develops in the medial end of the bony canal. Because the lateral end of the canal remains atretic or stenotic, the potential for cholesteatoma formation exists. Early in the development of this problem, symptoms such as pain or drainage from the ear canal or a fistulous track may be absent, and the diagnosis can only be made by CT.
Temporal bone CT is performed near the time of operation. Radiographic studies on infants are usually not recommended because the information is rarely applicable to immediate rehabilitative plans. In very young children, poor patient cooperation often necessitates anesthesia or results in a suboptimal study that must be repeated later. Almost all patients with cholesteatoma formation are older than 3 years, which is another reason to delay the CT evaluation until the patient is beyond that age (14).
MEDICAL MANAGEMENT
Unilateral Atresia
No immediate medical intervention is necessary in the infant discovered to have unilateral atresia, assuming there is normal hearing in the contralateral ear by ABR and OAE testing. The parents can be reassured that speech, language, and intellectual development will proceed normally. Preferential seating in school and possibly an FM system are advised, but a hearing aid is not recommended because of poor acceptance by most children. Many adults, however, find the consequences of unilateral hearing loss from atresia to be a significant aggravation at work and in social settings, and they more readily accept hearing aids. In patients with atresia of the ear canal, a bone-conduction hearing aid must be used. If the canal is only stenotic, an air-conduction aid is preferred because of cosmesis, improved sound localization (i.e., stimulation of one cochlea only), broader frequency response, less sound distortion, and comfort.
Bilateral Atresia
Early amplification within the first 3 to 4 months of life is essential in infants with bilateral atresia. Initial medical and audiologic evaluations can be completed within the first few months of life and a bone-conduction hearing aid or soft band BAHA can be fitted soon thereafter.
SURGICAL MANAGEMENT
Unilateral and Bilateral Atresia Repair
Although most otologic surgeons would consider atresia repair in bilateral cases, many are reluctant to operate on unilateral atresias. The issue is not the unilateral aspect of the hearing loss because most otologic surgeons will
explore the middle ear of a child with a large unilateral conductive hearing loss due to other causes (e.g., trauma, infection). The concern is the degree and predictability of hearing improvement that can be achieved, potential lifetime care of a mastoid cavity, and the risk to the facial nerve in atresia surgery. These concerns have prompted many surgeons to recommend delaying surgery in unilateral cases until adulthood, when patients can make their own decision based on the risks and benefits.
explore the middle ear of a child with a large unilateral conductive hearing loss due to other causes (e.g., trauma, infection). The concern is the degree and predictability of hearing improvement that can be achieved, potential lifetime care of a mastoid cavity, and the risk to the facial nerve in atresia surgery. These concerns have prompted many surgeons to recommend delaying surgery in unilateral cases until adulthood, when patients can make their own decision based on the risks and benefits.
An improvement in the hearing threshold to 25 dB or better eliminates the handicap of unilateral hearing loss. This degree of hearing improvement is not possible in all atresia patients, but it can be achieved in at least 50% of carefully selected patients. A mastoid cavity is not created if the “anterior” surgical approach is used, and risk to the facial nerve is minimized by understanding the abnormal development of this structure and by using intraoperative facial nerve monitoring. I and others contend that the benefits of binaural hearing and the possibility of achieving that goal are sufficiently great to offer corrective surgery to carefully selected children with unilateral atresia (10, 15).
Patients with bilateral atresia present less of a surgical dilemma. The goal in these cases is to restore sufficient hearing so that amplification is no longer needed. In contradistinction to ear selection for other otologic disorders, the “best” (as determined by CT evaluation) ear is selected for the initial surgical procedure. Most surgeons recommend operating as the child approaches school age and, depending on the hearing result, on the second ear within the next several years. As with many conductive hearing losses (e.g., otosclerosis), the possible use of amplification with a BAHA should be discussed. The probable need for yearly or semiannual cleaning of the reconstructed ear canal should also be acknowledged.
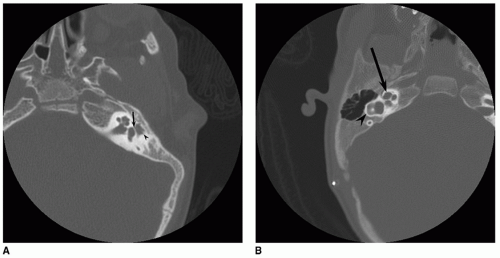 Figure 148.2 A: Aural atresia with mild middle ear dysplasia. Coronal CT shows an intact stapes with an intact incudostapedial joint (arrowhead), but the malleoincudal complex (arrow) is not properly formed. The tympanic segment of the facial nerve has a normal course. This patient would be a candidate for surgery. B: Aural atresia with severe middle ear dysplasia. Axial CT shows a diminutive middle ear cavity with no visible ossicles. The oval window is sclerosed (arrow), and the tympanic segment of the facial nerve is displaced laterally (arrowhead). This patient is not a candidate for surgery. |
Selection Criteria
Most patients undergoing atresia repair have a residual conductive deficit of at least 10 dB. Sensorineural function should be normal to achieve binaural hearing in unilateral cases or to obviate the need for a hearing aid in bilateral cases. Normal or near-normal sensorineural function in the contralateral ears is also important to avoid operating on the better hearing ear.
Although audiometric criteria can be defined quantitatively, the real art of patient selection is centered on the CT evaluation of the middle ear. Hypoplasia of the middle ear space, ranging from mild to severe, occurs in most cases of congenital atresia, and ossicular development can be expected to correlate directly with middle ear size. The risk of surgical complications will be minimized and the chances for a successful hearing result are increased if the middle ear and mastoid size are at least two-thirds of the normal size and if all three ossicles, although deformed, can be identified (Figs. 148.1 and 148.2). CT demonstration of the oval and round windows and a near-normal course of the facial nerve further define the ideal surgical candidate. The relationship of the facial nerve to the oval window (i.e., normally positioned or overhanging) and the position of the vertical segment are noted. Severe anterior displacement of
the vertical segment of the nerve restricts access to the middle ear space, reducing the chance for a successful hearing result and increasing the chance of facial nerve injury.
the vertical segment of the nerve restricts access to the middle ear space, reducing the chance for a successful hearing result and increasing the chance of facial nerve injury.
TABLE 148.1 JAHRSDOEFER GRADING SYSTEM FOR CONGENITAL AURAL ATRESIA | ||
|---|---|---|
|



