Congenital and Inherited Cataracts
J. Fielding Hejtmancik
Manuel B. Datiles III
Congenital cataracts are a significant cause of vision loss worldwide, causing approximately one third of blindness in infants. Roughly 8% to 25% of congenital cataracts are hereditary.1,2,3 Cataracts can lead to permanent blindness by interfering with the sharp focus of light on the retina and resulting in failure to establish appropriate visual cortical synaptic connections with the retina. Prompt diagnosis and treatment can prevent this. Understanding the biology of the lens and the pathophysiology of selected types of cataracts can yield insight into the process of cataractogenesis in general and provide a framework for the clinical approach to diagnosis and therapy.
Cell Biology of Cataracts
Transparency
The main functions of the lens are to transmit and focus light on the retina. Although about 80% of total refraction result from the cornea, the lens serves to fine tune the focusing onto the retina. The human lens is colorless when young, and a gradual increase in yellow pigmentation occurs with age.4 The lens efficiently transmits light with wavelengths from 390 nm to 1,200 nm. This range extends well above the limit of visual perception (approximately 720 nm). Lens transparency results from appropriate architecture of lens cells and tight packing of their proteins, resulting in a constant refractive index over distances approximating the wavelength of light.5,6 As lens proteins are diluted to concentrations below 450 mg/mL, light scattering actually increases.7,8 In addition, there is a gradual increase in the refractive index of the human lens from the cortex (1.38, 73% to 80% H2O) to the nucleus (1.41, 68% H2O), where there is an enrichment of tightly packed γ-crystallins.
Cataracts have multiple causes, but they are often associated with the breakdown of the lens’ microarchitecture,9,10,11,12 possibly including vacuole formation, which can cause large fluctuations in density, resulting in light scattering. In addition, light scattering and opacity will occur if there is a significant amount of high-molecular-weight protein aggregates 1,000 Å or more in size.13,14 The short-range ordered packing of the crystallins is important in this regard. For transparency, crystallins must exist in a homogeneous phase. The physical basis of lens transparency can be complex and has been reviewed elsewhere.5,13,14,15
Hereditary Cataract
Hereditary cataracts are estimated to account for between 8.3% and 25% of congenital cataracts.1,2 The lens alone may be involved, or lens opacities may be associated with other ocular anomalies such as microphthalmia, aniridia, other anterior chamber developmental anomalies, or retinal degenerations. Cataracts may also be part of multisystem genetic disorders such as chromosome abnormalities, Lowe syndrome, mitochondrial diseases, or neurofibromatosis type 2. In some cases, this distinction is blurred. Inherited cataracts may be isolated in some individuals and associated with additional findings in others, as in the developmental abnormality anterior segment mesenchymal dysgenesis, resulting from abnormalities in the PITX3 gene.16
Hereditary cataracts may be inherited as autosomal-dominant (most frequent), autosomal-recessive, or X-linked trait. Phenotypically identical cataracts can result from mutations at different genetic loci and may have different inheritance patterns, whereas phenotypically variable cataracts can be found in a single large family.17 Linkage analysis is a powerful tool used to sort out the different genetic loci that can cause human cataracts. Linkage studies emphasize the genetic heterogeneity of autosomal-dominant congenital cataracts. The number of known cataract loci has increased dramatically in the last few years to well over the 30 or so loci predicted to cause autosomal-dominant cataracts in man with no obvious sign that most loci have been identified. Obviously, much work remains to be done in understanding inherited congenital cataracts.
Lens Development and Cataracts
At birth, the human lens weighs about 65 mg. It grows to about 160 mg in the first decade of life and then more slowly to about 250 mg by 90 years of age.18 Proteins may constitute 60% of the total weight of the crystalline lens, much higher than most other tissues.19
The human lens is first anatomically visible at 3 to 4 weeks of gestation.20 The surface ectoderm over the eye field thickens into the lens placode and then invaginates toward the developing optic cup, forming the lens pit. The lens pit closes, and the resulting lens vesicle pinches off from the surface ectoderm.20 By the seventh week of development, cells forming the posterior layer of the optic vesicle begin to elongate and fill in the vesicle, loosing their nuclei. These become the primary fiber cells forming the embryonic lens nucleus.20 The remaining cells form the cuboidal anterior epithelium, some of which will divide, move laterally along the lens capsule and differentiate into secondary fibers.21 (Fig. 74.1).
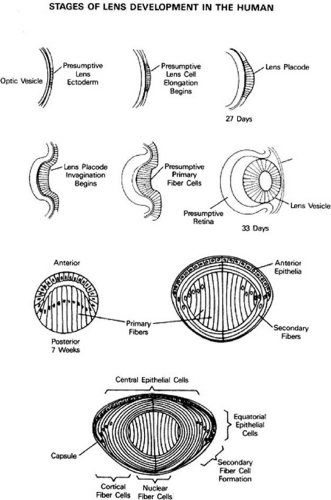 Figure 74.1 Development and structure of the crystallin lens. (Adapted from Piatigorsky J: Lens differentiation in vertebrates: A review of cellular and molecular features. Differentiation 19:134, 1981.) |
Although developmental control of lens differentiation is not yet well understood, Pax6, Rx, and a number of additional growth factors seem important for lens development.22,23,24,25,26 Mutations in Pax6 are associated with aniridia, which is often accompanied by cataracts,27 and Pax6 may co-operate with a number of additional factors including Sox2.()28 Six3, a vertebrate homologue of the Drosophila sine-oculis gene, can induce lens formation as well,29 and targeted deletion of Sox1 results in microphthalmia and cataract with failure of lens fiber cells to elongate.30 Pitx3, a member of the RIEG/Ptx gene family is expressed in the developing lens vessicle.31 A hereditary congenital cataract associated in some cases with the developmental abnormality anterior segment mesenchymal dysgenesis, can result from mutations in the PITX3 gene.16
The lens has a single layer of anterior epithelial cells overlaying the fiber cells wrapped onionlike around the lens nucleus.32 Cell division occurs in the germinative zone just anterior to the equator, and the cells then move laterally toward the equator, where the anterior epithelial cells begin to form secondary fibers. Both the anterior epithelial cells and fiber cells contain large amounts of crystallins. The anterior epithelial cells of the lens are connected by gap junctions,33 allowing exchange of low-molecular-weight metabolites and ions but have few or no tight junctions (zonula occludens), which would seal the extracellular spaces to low-molecular-weight proteins and ions.34,35 Ultrastructurally, anterior cuboidal epithelial cells are rich in organelles and contain large amounts of actin, myosin, vimentin, microtubules, spectrin, and α-actinin, which should stabilize them during accomodation.36,37,38 Differentiating lens fiber cells loose their organelles, including the mitochondria, Golgi bodies, and both rough and smooth ER. As the cells elongate, they move toward the lens nucleus. There is little extracellular space between the fiber cells, which have many interdigitations.21,32. Adjacent fiber cells are connected by many junctional complexes, which allow for intercellular passage of metabolites.37,38
The major soluble components of fiber cells are the lens crystallins, which make up approximately 90% of the water-soluble protein, and cytoskeletal components, including actin, myosin, vimentin, α-actinin, and microtubules.39 During this process, it seems clear that transcriptional control plays a significant role in the differential synthesis of lens crystallins.40 The distributions of the β-crystallins in chickens.41,42 and the β- and γ-crystallins in rats.43,44 provide examples of the spatial and temporal control of crystallin gene expression during lens development.
Classification of Congenital Cataracts
Characteristics used for diagnostic classification of human cataracts include age of onset, location, size, pattern, number, shape, density, progression, and severity in terms of interfering with visual acuity or visual function. They can also be categorized by presumed etiology. Roughly one third of congenital cataracts are associated with other disease syndromes and one-third are inherited, with the remainder being of unclear etiology.
Defined by age at onset, a congenital or infantile cataract is visible within the first year of life, a juvenile cataract occurs within the first decade of life, a presenile cataract occurs before the age of 45 years, and the so-called senile or age-related cataract, thereafter. The age of onset of a cataract does not necessarily indicate its etiology. Congenital cataracts may be hereditary or secondary to a noxious intrauterine event. Cataracts associated with a systemic or genetic disease may not occur until the second or third decade (e.g., cataracts associated with retinitis pigmentosa). Even age-related cataracts, thought to be due to multiple insults accumulated over many years, have a genetic component, making certain individuals more vulnerable to the environmental insults.
There are several classification systems that have been developed based on the anatomic location of the opacity. In an attempt to deal with congenital cataract, Merin has proposed a system based on morphological classification. Accordingly, the cataract is classified as total (mature or complete), polar (anterior or posterior), zonular (nuclear, lamellar, sutural), and capsular or membranous.45
As discussed earlier, because lens development follows a well-documented timed sequence, the location of a lens opacity provides information about the time at which the pathological process intervened, thereby aiding in determining the etiology. Nuclear opacities from the most central region outward denote cataract formation occurring at the time of the development of that portion of the involved lens nucleus—embryonic (first 3 months), fetal (third to eighth month), infantile (after birth), or adult. Because the lens fibers are laid down constantly throughout life, lens opacities that develop postnatally present as cortical opacities or appear just beneath the posterior lens capsule as subcapsular opacities, for example, cataracts caused by topical steroid drugs and radiation.
Polar opacities involve either the anterior (Fig. 74.2A) or posterior (Fig. 74.2B) pole of the lens and may include the posterior subcapsular lens cortex (Fig. 2C) extending to the lens capsule. Posterior subcapsular cataracts can also occur secondarily to a variety of insults. Although they have been associated with proliferation of dysplastic bladderlike fiber cells called Wedl cells, at least some posterior subcapsular cataracts appear to be due to abnormalities of the posterior fiber ends.46 When both anterior and posterior poles are involved, the term bipolar is used. Isolated anterior polar cataracts are usually small, bilateral, and nonprogressive and do not impair vision. They may be inherited as an autosomal-dominant trait,47 or may be associated with microphthalmos, persistent pupillary membrane, or anterior lenticonus. Posterior polar cataracts also may be inherited as a dominant trait.48 or may be sporadic and unilateral, and they can be associated with abnormalities of the posterior capsule, including lentiglobus or lenticonus or with remnants of the tunica vasculosa. Although they are usually stable over time, they may progress and can be associated with capsular fragility.
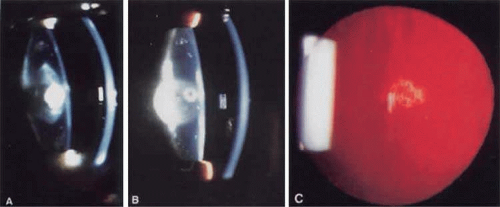 Figure 74.2 Examples of polar cataracts. A. A dense anterior polar cataract visible on slit lamp examination. Some opacification of the lens nucleus is also visible. B. A dense posterior polar cataract is visible on slit lamp examination. A smaller anterior polar cataract is also visible so that this would be termed a bipolar cataract. C. Posterior subcapsular cataract. |
Nuclear cataracts show opacities in the fetal or fetal and embryonic lens nucleus (Fig. 74.3A,B). They can show a wide variation in severity, from dense opacities involving the entire nucleus to pulverulent (or dusty-appearing) cataracts involving only the central nucleus or discrete layers (see later discussion).
Lamellar cataracts (Fig. 74.3C,D) affect lens fibers that are formed at the same time, resulting in a shell-like opacity at the level at which the fibers were laid down at the time of the presumed insult. They are the most common type of congenital cataract and may be inherited in a dominant fashion.49,50,51,52,53 Some cataracts have associated arcuate opacities within the cortex called cortical riders.
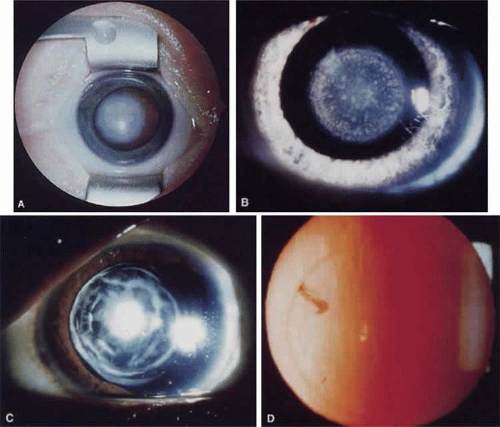 Figure 74.3. Examples of nuclear and lamellar cataracts. A. A dense nuclear cataract. The macula and optic nerve are obscured by this cataract. B. A punctate nuclear cataract. C. A multi-lamellar cataract with an anterior polar component. D. A very fine nuclear lamellar pulverulent cataract viewed by retroillumination. A rider is visible at about 10 o’clock. |
Sutural or stellate cataracts (Fig. 4A,B) affect the regions of the fetal nucleus on which the ends (or feet) of the lens fibers converge, called the Y sutures. These are visible by slit-lamp biomicroscopy as an upright Y anteriorly and an inverted Y posteriorly, even in normal lenses. Theories of cataract development.54 suggest that abnormalities in lens-fiber development or maturation may lead to a predisposition to cataract development later in life. This is supported by examples in animals (the Philly mouse) and in humans (gyrate atrophy).55 Sutural cataracts can also be inherited as autosomal-dominant traits.49 Cerulean, or blue dot cataracts, are characterized by numerous small, bluish opacities in the cortical and nuclear areas of the lens (Fig. 74.4B).53
 Figure 74.4. Examples of sutural or stellate cataracts. A. A sutural cataract with a nuclear lamellar component B. A sutural cataract with a cortical cerulean or blue dot component. |
Mature or total cataracts may represent a late stage of any of the above types of cataracts in which the entire lens is opacified (Fig. 74.5A). Membranous cataracts result from resorption of lens proteins, often from a traumatized lens, with resulting fusion of the anterior and posterior lens capsules to form a dense white membrane. They usually cause severe loss of vision. Other varieties of cataract can usually be described through a combination of the previously mentioned terms, although there are some specialized cataracts that have unique characteristics, such as the ant’s egg cataract (Fig. 74.5B,C), in which a mutation in connexin 46 causes beaded structures to form from the lens.56,57
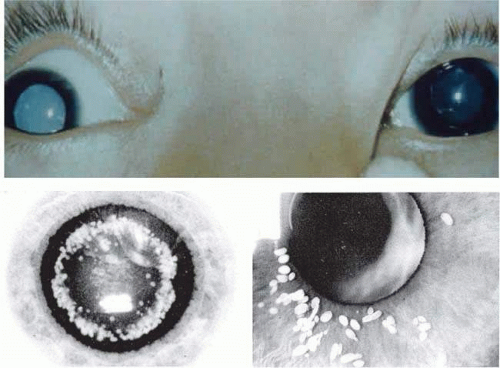 Figure 74.5. A. Example of total or mature cataract. B–C. Examples of ant’s egg cataract, courtesy of R. Riise and Lars Hansen. |
Molecular Biology of Cataracts
Lens Crystallins
Crystallins can be defined as proteins that are found in high concentration in the lens. They make up more than 90% of water-soluble lens protein and fulfill a critical structural role for transparency and refraction.19 Classically, the ubiquitous crystallins, found in all species, can be separated into three soluble and one insoluble fraction.58 The soluble fractions comprise the α-, β-, and γ-crystallins, found in all vertebrate lenses (Table 74.1). In the mature human lens, α-crystallins make up 40%, β-crystallins 35%, and γ-crystallins 25% of total crystallin protein. The β- and γ-crystallins show sequence and tertiary structure homology and form the βγ-superfamily (see later discussion).
α-Crystallins
The α-crystallins are products of two similar genes, αA- and αB-crystallin. The sequences of human αA- and αB-crystallins are 57% identical,59,60,61 with αA-crystallin containing 173 and αB-crystallin 175 residues, both having a predominantly β-sheet structure.62 Native α-crystallins exist in the lens as globular complexes ranging from 300 to 12,00 kDa. Currently, the most favored structural.63 model proposes that the α-crystallin aggregate behaves as a protein micelle.64,65 It can be shown that αA and αB occupy equivalent and dynamic positions in the aggregate, with subunit exchange occurring easily.65,66,67,68 Cryo-electron microscopy has shown that recombinant αB-crystallin has a hollow central core surrounded by a protein shell with variable monomer packing.69 Although αA-, αB-, and even αAins-crystallins appear to occupy equivalent positions in the α-crystallin aggregate,65,66 they are expressed in different tissues, have radically different effects in knockout mice,70,71 and differ in their phosphorylation,72,73 structural properties,74 and chaperone functions.74, suggesting that each fulfills a unique role in the lens.
Both αA- and αB-crystallin can function as molecular chaperones in that they can protect both β and γ-crystallins and enzymes from thermal aggregation. However, they do not cycle these proteins in the manner of true chaperones,75,76 even though there is some evidence that αB-crystallin binds ATP specifically.77 The chaperone function of the α-crystallins probably serves to protect lens proteins from denaturing with age and could explain their presence in nonlenticular tissues. It involves the C-terminal domain of the protein,78 which participates in structural transitions resulting in the appropriate placement of hydrophobic surfaces within a multimeric molten globular state,79 perhaps with hydrophobic and hydrophilic regions bound by discrete parts of the α-crystallin protein.80 The chaperone function of the α-crystallins should serve to protect against cataractogenesis by reducing the aggregation of partially denatured proteins that accumulate within the lens during aging. αB-Crystallin and, to a lesser extent, αA-crystallin are expressed in tissues outside the lens.81 In this fashion, the α-crystallins are similar to enzyme-crystallins and may have important metabolic functions in the lens and other tissues.
Mutant or absent α-crystallins have been associated with inherited cataracts. Autosomal-dominant congenital zonular, nuclear, fan-shaped, and polar cataracts have been associated with mutations of αA-crystallin on chromosome 21q22.3 (Table 74.2). A presenile lamellar cataract progressing to total cataract has been associated with a G98R mutation, and an autosomal-recessive total cataract with microcornea and punctate lenticular opacities in carriers has been associated with an R54C mutation. Interestingly, all cataractogenic mutations identified in CRYAA to date involve arginine residues or are truncations, suggesting that these residues might be particularly critical for α-crystallin stability or chaperone activity. Mutations in αB-crystallin can cause autosomal-dominant cataracts (Table 74.2), at least one of which acts through a dominant-negative mechanism on αB-crystallin’s chaperone function.82 However, when arginine 120 is changed to glycine (R120G), it forms large aggregates with desmin in smooth muscle cells, causing a severe myopathy associated with mild discrete cataracts.83 This is reminiscent of the behavior of αB-crystallin in αA-crystallin knockout mice,70 whereas αB-crystallin knockout mice develop a late-onset fatal myopathy without cataracts.71
Table 74.2. Mapped Mendelian cataract loci and mutations. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
βγ-Crystallins
The β- and γ-crystallins are antigenically distinct but are members of a related βγ-crystallin superfamily, as determined by sequence conservation of 30%,84 and conserved tertiary structure of their central globular domains.85,86 They differ with respect to their developmental expression and association of the β-crystallins but not the γ-crystallins into macromolecular complexes.
γ-Crystallins
The γ-crystallins have molecular weights of about 21 kDa and show the highest symmetry of any crystallized protein, which may contribute to their high stability in the lens. The structures ofγB-, γD-, γE-, and γF- crystallin have been determined and are very similar.87,88,89,90 The amino acids of the core domains are arranged into four repeated segments called “Greek key” motifs. Each Greek key motif consists of an extremely stable, torqued β-pleated sheet resembling the characteristic pattern found on classical Greek pottery.87 The first and second motifs are in the N-terminal domain, and the third and fourth motifs are in the C-terminal domain of the protein.
The γ-crystallins accumulate specifically in the lens fibers and are the predominant crystallins in the lens nucleus, which maintains the highest protein concentration and is the least hydrated section of the lens. Thus, γ-crystallins appear especially adapted for high-density molecular packing.89 γ-Crystallins can be subdivided into two groups, γABC- and γDEF-crystallins.91,92 Proteins in the latter group have higher critical temperatures for phase separation and are largely responsible for the occurrence of the “cold cataract,”.93 a reversible opacity, which occurs on cooling of the lens.90 βγ-Crystallins appear distantly related to protein S, a sporulation-specific protein of the bacteria Myxococcus xanthus, to spherulin 3a of the slime mold Physarum polycephalum,94 to CRBG-GEOCY of the sponge Geodia cydonium,95 and to A1M1, a tumor suppressor gene.96 These microbial proteins can be induced by physiological stresses such as osmotic stress,94 providing a functional parallel to the α-crystallins and some taxon-specific crystallins (see later discussion).
γS-crystallin (formerly called βS-) represents a link between the β- and γ-crystallins.97,98,99,100 Many physical and chemical properties of the γS protein resemble those of β-crystallins.101,102 γS-Crystallin is also expressed later in development than the other γ-crystallins, especially in the adult when expression of other γ-crystallins is low or has ceased,103 and is expressed in birds and reptiles.104,105 However, in contrast to the β-crystallins and like the other γ-crystallins, γS-crystallin exists in solution as a monomeric protein. It is especially important that the gene structure of γS-crystallin has three exons,98,99 making it similar to the other γ-crystallins and distinctly different from the β-crystallins, which are encoded by genes with six exons.19 However, while most γ-crystallin genes are clustered on chromosome 2, the γS-crystallin gene is found alone on chromosome 3.86
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


