65 Congenital Abnormalities of the Head and Neck • 1/7000 to 8000 live births • Common cause of neonatal respiratory distress • 50 to 60% unilateral • 29% pure bony, 71% mixed bony and membranous • Female > male (2:1) • Autosomal recessive inheritance • Gene chromodomain 7 • Failure of breakdown of buccopharyngeal membrane on day 45 of gestation • Neural crest migration failure = another theory • Common association with other anomalies: – C—coloboma (eye defects) – H—heart defects – A—choanal atresia (nasal blockage) – R—retardation of growth and developmental delay – G—genitalia (small penis, undescended testicles) – E—ear anomalies—malformed pinnae, hypoplastic incus, cochlear anomalies, absent Sccs • Respiratory distress at birth • Infant crying relieves obstruction • When neonate closes mouth pattern of cyclic obstruction develops leading to respiratory failure • Unilateral cases present later in life with unilateral rhinorrhoea • Obligate nose breathers until 6 to 9 months age • Confirmed by inability to pass ⅚ Fr gauge feeding tube • Endoscopic examination • CT scan determines type of obstruction and thickness • ECHO • Transnasal correction in 1st week of life • Unilateral can be corrected at 1 year • Curette partition • Use 3.5 mm ETT as stent • Use posterior membranous flaps to cover bone • Complications: • CO2 laser, endoscopically guided, Nd:YAG laser = alternatives • Superior visualization • Impaired palatal growth a potential problem in neonate—suitable only for >5 years old • Pharyngeal segments of primitive notochord remain connected to endoderm in nasopharynx • Bursa located in midline of nasopharynx • If bursa occluded by inflammation → Thornwaldt cyst—can become infected • Anterior to invagination and above bursa is a small pharyngeal hypophysis developed from Rathke pouch—sometimes persists as craniopharyngeal canal running from sella turcica through body of sphenoid • Appear clinically in 2nd to 3rd decades • Male = female • Intermittent/persistent postnasal discharge (tenacious mucus/purulent material) • Associated with odynophagia, halitosis, dysgeusia, dull occipital headache • Midline cystic mass superior to adenoids seen on endoscopy • Cyst lined by respiratory epithelium • CT/MRI—well defined • Excision or wide marsupialization • Lined by columnar or cuboidal ciliated epithelium • May become infected or rupture intracranially • Commonly associated with galactorrhea, visual field loss, and hypopituitarism • Transsphenoidal cyst drainage and biopsy of the wall required • Rare malignant neoplasms arising from notochordal remnants • 5th to 6th decade of life • 50% in spheno-occipital area • Male = female • Expanding nasopharyngeal mass • Frontal headaches • CN palsies (VI n in 60%) • Pituitary abnormalities • Children <5 years have wider range of presenting symptoms • CT/MRI to investigate and plan surgery • Surgical resection via skull base approach • 5-year survival = 50% • Radiotherapy increases survival rates • Chemotherapy indicated if mets occur • Arise from Rathke pouch • Located in sellar, parasellar, and III ventricle regions • Composed of well-differentiated epithelial elements, e.g., cysts, ameloblasts • 10 to 15% of all childhood intracranial neoplasms • Visual field defects, sudden blindness, extraocular motor paralysis, and hypopituitarism • Median age = 7.5 years • Male = female • Surgical resection followed by radiotherapy • 10-year survival = 90% • Surgical removal has high risk of death endocrinologic complications, and behavioural dysfunction • Those located in an enlarged sella can be removed transsphenoidally • 1/700 births • Increased risk if older sibling affected • Teratogens: ethanol, retinoids, folate antagonists • Maternal smoking also causative • Syndromic or nonsyndromic • Cleft lip is due to failure of fusion between medial nasal, maxillary, and lateral nasal prominences • Cleft lip either complete (muscular diastasis [separation] of orbicularis oris) or incomplete • Cleft palate formation is hypothesized to result from: • Primary palate: lip, alveolar arch, palate anterior to incisive foramen (premaxilla) • Secondary palate: soft palate and hard palate posterior to incisive foramen • See Fig. 65.1 • Alone, feeding is not normally a problem • Normally repaired at 2–6/12 of age
65.1 Choanal Atresia
65.1.1 Epidemiology
65.1.2 Aetiology
 CHARGE:
CHARGE:
 Branchial abnormalities
Branchial abnormalities
 Humeroradial synostosis
Humeroradial synostosis
 Mandibular facial synostosis
Mandibular facial synostosis
 Microcephaly
Microcephaly
 Micrognathia
Micrognathia
 Nasopharyngeal abnormalities
Nasopharyngeal abnormalities
 Palatal defects
Palatal defects
65.1.3 Symptoms
65.1.4 Investigations
65.1.5 Treatment
Transnasal Approach
 Meningitis
Meningitis
 CSF leak
CSF leak
 Brain injuries
Brain injuries
 Gradenigo syndrome
Gradenigo syndrome
 Cervical vertebral subluxation
Cervical vertebral subluxation
 Restenosis
Restenosis
Transpalatal Approach
65.2 Thornwaldt Cyst and Rathke Pouch Cyst
65.2.1 Embryology/Pathology
65.2.2 Thornwaldt Cysts
65.2.3 Rathke Pouch Cysts
65.3 Chordomas
65.3.1 Craniopharyngiomas
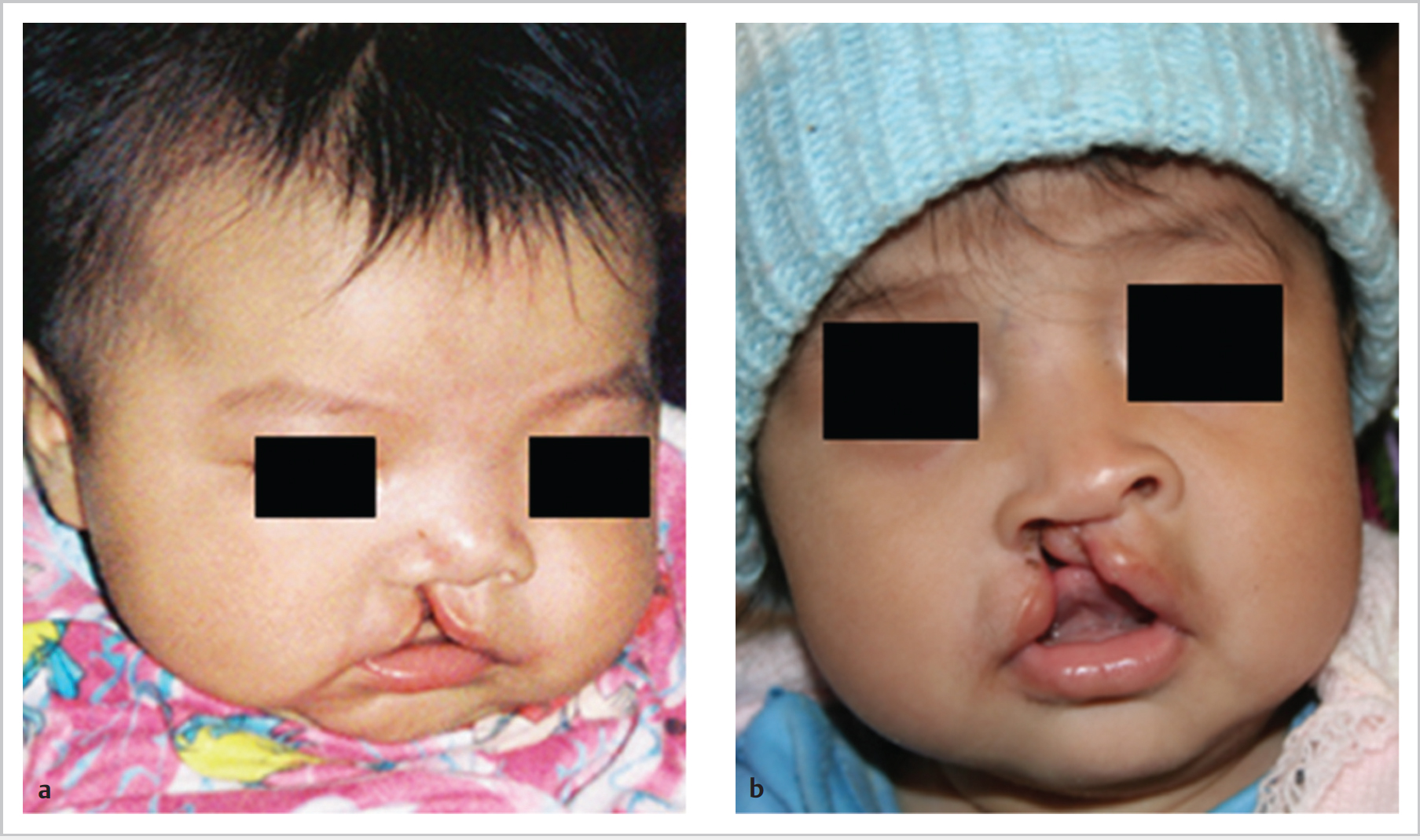
65.4 Cleft Lip and Palate
65.4.1 Epidemiology
 Over 20 syndromes associated, e.g., Apert, Treacher–Collins, Pierre Robin, and DiGeorge
Over 20 syndromes associated, e.g., Apert, Treacher–Collins, Pierre Robin, and DiGeorge
65.4.2 Aetiology
 Defects in palatal shelf growth
Defects in palatal shelf growth
 Delayed shelf elevation
Delayed shelf elevation
 Failed shelf elevation
Failed shelf elevation
 Defective shelf fusion
Defective shelf fusion
65.4.3 Features and Symptoms Cleft Lip
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


