Fig. 5.1
Cochlear lateral wall and potassium ion recycling. The ionic composition with high potassium concentration and endocochlear potential are maintained by ion transport through the cochlear lateral wall. Potassium ion (K+) influx from endolymph into hair cell is evoked by sound stimuli, followed by secretion of ion to extracellular space in the organ of Corti. K+ are transferred through the epithelial gap junction network to the cochlear lateral wall, where the connective tissue gap junction network and the stria vascularis recycle and secrete K+ into endolymph in the scala media. The area of each type of fibrocytes in the spiral ligament is indicated by I–V.
The fibrocytes in the spiral ligament form a network of ion-transporting connective tissue cell gap junction system that facilitates recycling of K+ from perilymph back to endolymph. Spiral ligament fibrocytes are classified into five types according to characteristics in morphology and immunohistochemistry [14]. Type I fibrocytes are distributed between the stria vascularis and the bony wall of the cochlea and stained for carbonic anhydrase (CA) isozymes II and III and CK isozyme BB. Type I fibrocytes have few cellular processes and contained few cellular organelles in contrast to type II fibrocytes which possessed abundant mitochondria. Type II fibrocytes lie under the outer sulcus and spiral prominence and possess abundant cellular processes. Type II fibrocytes express ion transporters including Na+,K+-ATPase and Na+,K+,2Cl− cotransporter, suggesting that type II fibrocytes are involved in regulation of the solute content in the cochlear lateral wall. Type III fibrocytes are located in the area adjacent to the cochlear bony wall in the inferior region of the spiral ligament. Type III fibrocytes contained not only CA II and III and CK isozymes BB and MM but also various types of cytoskeletal filament including actin, which indicates that type III fibrocytes play roles in metabolism as well as mechanical support for the basilar membrane. Type IV fibrocytes are placed facing to the scala tympani in the inferior part of the superficial spiral ligament. Type IV fibrocytes express Na+,K+-ATPase, CA II and III, and CK. Type V fibrocytes reside in the suprastrial area between the scala vestibuli and the cochlear bony wall. Type V fibrocytes appear heterogenous in terms of expression of ion transporter, the more superficial having predominantly ATPase and the deeper only expressing CA; however, morphological characteristics of type V fibrocytes resemble that of type II fibrocytes. Types II, IV, and V fibrocytes function to pump K+ from the perilymph and produce a K+ flow to type I fibrocytes which are electrically connected to the basal cells in the stria vascularis [15].
The stria vascularis is made up of three layers of cells: marginal cells in the luminal surface to the scala media, the intermediate cells, and basal cells adjacent to the spiral ligament (Fig. 5.2). The marginal cells are a layer of polarized epithelial cells that are derived from the epithelium of the cochlear duct and form the luminal surface of the scala media. Marginal cells are abundant in cytokeratin proteins and also include several molecules associated with ionic plumps and channels. The intermediate cells, which are probably derived from the neural crest, contain melanin and are referred to as melanocytes [16]. When melanocytes are missing in the stria vascularis, EP is not generated and hearing is severely impaired [17]. Basal cells are located lateral to the intermediate cell layer. These flat cells form a continuous layer and a dense network of gap junction complexes with neighboring basal cells. It is not completely clear whether these cells are derived from mesodermal or neural crew origins.
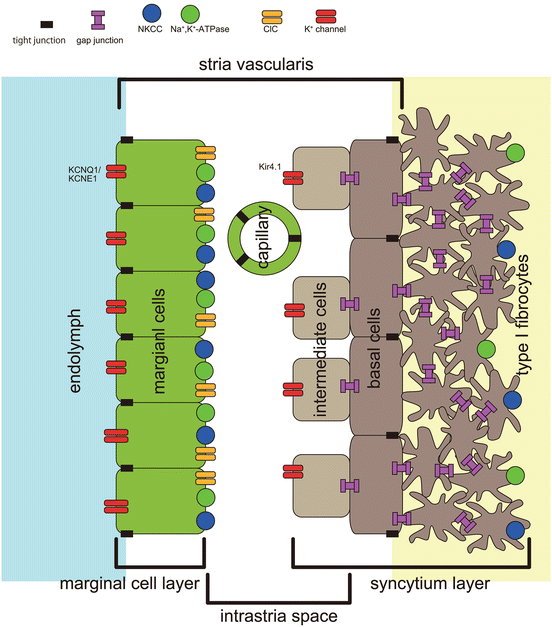
Fig. 5.2
Schematic of the two-pump model in the stria vascularis. Ion transporters and channels and three cell layers in the stria vascularis contribute generation of endocochlear potential and high potassium ion (K+) concentration in endolymph. Intermediate cells, stria basal cells, and type I fibrocytes in the spiral ligament are electrically connected by gap junction. While stria intermediate cells express Kir4.1, K+ channel, marginal cells express KCNQ1/KCNE1, K+ channel, in the apical membrane. Tight junction blocks extracellular ion exchange between neighboring compartments both in the marginal cell layer and the basal cell layer. NKCC, Na+,K+, 2Cl− cotransporter; ClC, ClC-K/Barttin-type Cl− channel
At the lateral extremity of the epithelial cell gap junction system, root cells reside in partwithin the outer sulcus region. The root cells are named after characteristic fingerlike projection from their cell bodies and infiltrate between the mesenchymal fibrocytes of the spiral ligament. They display a graded variation of their gross morphological properties along the tonotopic axis of the cochlear spiral, particularly in terms of the number and size of the root processes. Also, the root cells possess the functional specialization including intercellular tight junction and gap junction [18, 19].
Melanocytes have been known to reside as intermediate cells of the stria vascularis. Intermediate cells express Na+,K+-ATPase and K+ channels which are essential for production of EP. Intrastrial space is quite rich in blood vessels. The vessel wall constitutes the intrastrial fluid-blood carrier of specialized endothelial cells, which are surrounded by pericytes and melanocytes. Perivascular melanocytes in the intrastrial space maintain barrier integrity by controlling tight junction and adherence junction protein expression [20].
Bone marrow-derived cells also are widely distributed in the cochlear lateral wall [21, 22], most of which have phenotypes as tissue-resident macrophages. Specific roles of cochlear resident macrophages are largely unknown; however, macrophages play roles in protection of hair cells following application of ototoxic drugs or innate immune systems in the cochlea [23, 24]. In addition, Hirose et al. reported that bone marrow-derived cells are attracted to the cochlear sensory epithelium when the cochlea is insulted by acoustic overstimulation [25]. These observations suggest that bone marrow-derived cells play substantial roles in maintenance of cochlear function and homeostasis.
5.3 Physiology of the Cochlear Lateral Wall
The cochlea is the organ that continuously controls water and ion homeostasis for auditory perception. Like the retina, cochlea maintains continuous extracellular potential and ion gradients through transporting ions even in the absence of an acoustic stimulus, which was called “silent current” by Zidanic and Brownell [26]. In addition, these gradients are not generated by hair cells but in the stria vascularis, which benefits significantly for the detection of mechanical stimuli. As generation of EP and K+ recycling from perilymph to endolymph are inseparably connected, we will discuss physiological roles and mechanisms of the cochlear lateral wall in terms of generation of EP.
As described above, the cochlear endolymph exhibits a positive potential of +80 mV, which is called the endocochlear potential. von Bekesy in 1952 first reported the DC potential in endolymph with a +80 mV positive potential to the perilymph [27]. Davis et al. measured the cochlear microphonic and found that the energy of the cochlear microphonic is derived from the polarization potential across the interface between the endolymph and the interior of the hair cells, which is called “battery theory.” In addition, they described that the movements of hair bundles modulate the flow of electric current [28]. These batteries that drive auditory-evoked excitation in hair cell are comprised of electrochemical gradients across the apical surface of hair cell produced by EP. Since then, so many works have been done on the mechanism of generation of EP and high potassium concentration [29, 30]. Salt et al. found the intrastrial space with a positive potential and a low K+ concentration through measurement of the potential and K+ gradient in the stria vascularis using K+-selective electrodes. The results also indicate that EP is not generated by the marginal cells alone but also by K+ movement across the cell membrane of components in the stria vascularis [31]. The works by Salt et al. redefined the investigation for EP as research on cellular compartments and ion pumps that positively transport cation and anion across cellular membrane and intercellularly.
The two-cell model, also referred to as the five-compartment model [32], has been widely accepted as the mechanism for origin of EP [33]. In this hypothesis, the cochlear lateral wall comprises two epithelial compartments, the strial marginal cells and the syncytium that forms cellular complex of fibrocytes, strial basal cells, and strial intermediate cells. Type I fibrocytes, strial basal cells, and strial intermediate cells are interconnected by gap junction network, which means these cells are electrically equivalent. Type I fibrocytes in the cochlear lateral wall express both Na+,K+-ATPase and Na+,K+,2Cl− cotransporter. Vascular perfusion of either ouabain, an inhibitor for Na+,K+-ATPase, or furosemide, an inhibitor for Na+,K+,2Cl− cotransporter, dramatically reduces EP [34, 35], indicating that these two ion transporters are required for generation of EP. The strial intermediate cells express Kir 4.1 [36] which secretes K+ into the intrastrial space. Vascular perfusion of Ba2+, a potential blocker for the Kir family, remarkably suppresses EP [37], suggesting that the K+ diffusion potential is primarily formed by Kir4.1 and critical for generation of EP. Tight junctions between n the basal cells make the syncytium a diffusional barrier and serve as the boundary of the apical surface composed of intermediate cell membranes and the basolateral surface comprising the fibrocyte membranes. Mice lacking Claudin11, a major constituent of tight junctions of basal cells, exhibit disruption of EP despite normal K+ recycling, indicating the importance of basal cell tight junctions in the stria vascularis for generation of EP [38, 39]. The results also support two-cell model that EP is not generated by the marginal cells alone but the intrastrial space contributes generation of endocochlear potential. Expression of Na+,K+-ATPase and Na+,K+,2Cl− cotransporter in the basolateral membrane of the strial marginal cells sustains a low K+ concentration in the extracellular fluid in the intrastrial space. Chloride ions exit from the marginal cells through ClC-Kb chloride channels, Barttin, which is supposed to be essential for K+ recycling in the stria vascularis [40]. KCNq1/KCNE1 K+ channels which are expressed in the apical surface of the marginal cells secrete K+ into the endolymph contributing a K+ diffusion potential and the dynamics of EP [41]. Whereas the basal cells in the stria vascularis express only Claudin11, the marginal cells express a variety of Claudin family including Claudin1, 2, 3, 8, 9, 10, 12, 14, and 18, suggesting importance of the barrier formation in the marginal cells [42]. Researches in the field of physiology and molecular biology have made substantial advance to complete understanding for roles and mechanisms of the cochlear lateral wall. In the next section, we will discuss potential molecular targets for regenerative medicine in the cochlear lateral wall identified in either humans and mice or both.
5.4 Molecular Basis of the Function of Cochlear Lateral Wall
Several genes that are involved in ion transporting are reported to account for hereditary hearing loss. Mutations in GJB2 encoding connexin 26 are the most prevalent inherited source of congenital hearing loss in humans [43]. In most of these cases, the inheritance type is autosomal recessive; however, some cases of dominant inheritance are also reported. In Gjb2 knockout mice, degeneration of the organ of Corti is observed as early as postnatal day (P)14 and their hearing are profoundly impaired [44]. Endolymphatic K+ concentration and EP are much lower in Gjb2 knockout mice than wild type, suggesting that Cx26 is essential for maintenance and function of the organ of Corti, but is not required for normal development of the cochlear sensory epithelium. A Gjb6 knockout mouse model was also developed, and homozygous mice are hearing impaired and lack EP. Degeneration of the organ of Corti is observed from P18, which is similar to Gjb2 mutant mice [45].
The SLC26 family is responsible for membranous transporting of anion, including chloride, iodide, and bicarbonate. SLC26A4 mutations are associated with syndromic hearing loss, Pendred syndrome, and non-syndromic hereditary hearing loss [46]. Most of the patients display radiologically detectable structural malformations of the inner ear, the most common feature of which is an enlarged vestibular aqueduct. Enlarged endolymphatic ducts are also observed in some patients with non-syndromic hereditary hearing loss due to SLC26A4 mutations. In the mouse inner ear, Slc26a4 is expressed on apical surface of cells covered by the endolymphatic space [47]. Slc26a4 knockout mice exhibit waltzer-like vestibular dysfunction and complete deafness with a severe dilatation of the endolymphatic duct and sac [48]. Functional analyses revealed that Slc26a4 knockout mice gradually loss EP, beginning at P12, before normal onset of hearing despite normal concentration of K+ in the endolymph.
In the cochlea, the essential segregation of endolymph from perilymph is achieved by tight junctions of epithelial cells bordering the fluid compartments. Tight junctions are composed of at least three types of transmembrane proteins: occludin, claudins, and the junction adhesion molecule family. Claudin proteins are widely expressed in the inner ear, with 10 types of claudin proteins of differential distribution or localization [42]. Recessive mutations of human CLDN14 were identified as a source of non-syndromic hearing loss [49]. Claudin14 is detected in tight junctions of the reticular lamina in the organ of Corti in mice, and Claudin14-null mice are deaf, whereas their EP stays normal [50]. Tight junctions in the basal cells of stria vascularis are primarily composed of Claudin11, and Claudin11-deficient mice show severe hearing loss without obvious degeneration of the organ of Corti [39]. In addition, EP in Claudin11-deficient mice is suppressed, while K+ concentration in the endolymph is maintained at almost normal level. These findings indicate that establishment of the basal cell barrier in the stria vascularis is indispensable for hearing function through generation and maintenance of EP.
Kcne1, Kcnq1, and Kcnq4 encode for subunits of low-voltage-activated K+ channel, which are the major determinants of cellular depolarization in excitable cells. Stria vascularis marginal cells secrete K+ into the endolymph by K+ channel composed of Kcnq1 and Kcne1 subunits. Mutations in KCNE1 or KCNQ1 in human induce syndromic hearing loss with cardiac symptoms, including prolonged QT interval in the electrocardiogram and arrhythmias (Jervell and Lange-Nielsen syndrome) [51–53]. Kcne1 or Kcnq1 knockout mice exhibit severe hearing loss and vestibular dysfunction [54, 55]. Although morphology of the cochlear duct is likely to be normal at birth, degeneration develops later after birth including a collapse of Reissner’s membrane and a decrease in the volume of endolymphatic space. Kcnq4 is detected in the basolateral membrane of cochlear hair cells, suggesting that Kcnq4 channels are responsible for secretion of K+ from hair cells to the perilymph [56].
Kir4.1, or KCNJ10, is expressed in the intermediate cells of stria vascularis [57]. K+ concentration as well as EP and volume of the endolymphatic space is reduced in Kcnj10 knockout mice, suggesting that the Kir4.1 channel provides the molecular mechanism for generation of EP in concert with other channels for K+ secretion, as is indicated by the fact that Slc26a4 knockout mice lack Kir4.1 expression in the stria vascularis [58].
In human, Bartter syndrome IV is an autosomal recessive disorder characterized by congenital deafness and severe renal salt and fluid loss, which is caused by mutations in Barttin, a beta-subunit of ClC-Ka and ClC-Kb chloride channels [59]. NKCC, Na+,K+,2Cl− cotransporter, and ClC, Barttin-type Cl− channel are expressed in the basolateral membrane of the stria marginal cells. Barttin-knockout mice demonstrate severe hearing loss with a decrease of EP despite normal concentration of K+ in the endolymph [60]. These observations indicate that Cl− transport in the stria vascularis is also involved in the formation of EP.
Finally, DFN3, an X chromosome-linked non-syndromic mixed deafness, is caused by mutations in the POU3F4 gene, which encodes a POU transcription factor [61, 62]. Pou3f4-deficient mice were created and found to exhibit profound deafness with a dramatic reduction in EP. Histological analyses demonstrated a hypoplasia of fibrocytes in the cochlear lateral wall. The findings suggest that fibrocytes responsible for K+ homeostasis in the lateral wall play a critical role in generating EP and auditory function as well. Taken together, these molecules described above could be potential targets for regenerative medicine to improve hearing in disorders of the cochlear lateral wall through regenerative therapy.
5.5 Targets of Regenerative Medicine
In human, some types of hearing impairment are clinically suggested to be caused by disorder or damage of the cochlear lateral wall. The most widely referenced scheme for describing age-related hearing loss is one attributed to Schuknecht, in which three major cochlear structures, afferent neuron, organ of Corti, and stria vascularis, can degenerate independently [63, 64]. In addition, an age-related loss of fibrocytes in the spiral ligament has been reported in the basal portion of the mouse cochlea [65]. Moreover, Ohlemiller et al. reported that degeneration of the strial intermediate cells plays essential roles in age-related hearing loss through providing melanin to both the marginal cells and the basal cells [66]. Taken together, the cochlear lateral wall is very likely to be affected in the pathology of presbycusis according to the data shown above.
Pathological changes in fibrocytes have been linked to noise-induced hearing loss as well as age-related hearing loss, where fibrocyte degeneration was shown to precede loss of hair cells and neurons [65]. Widespread degeneration of fibrocytes in the spiral ligament, especially among the type IV fibrocyte areas, was observed in aged mouse cochlea prior to hair cell loss or degeneration of spiral ganglion neurons, suggesting that pathological changes in the fibrocytes in the cochlear lateral wall might be responsible for hair cell degeneration in age-related hearing loss.
Sudden deafness, or sudden-onset unilateral hearing loss due to unknown etiology, is suggested to be caused, at least in part, by damage of the lateral wall. Whereas some studies focused on degeneration of the spiral ganglion or the organ of Corti in models of transient cochlear ischemia [67], recent studies have revealed that the cochlear lateral wall is also affected by cochlear ischemia or energy failure, which seems to be reasonable as vasculature and mitochondrial activity are abundantly distributed in the cochlear lateral wall. In a mouse model of acute mitochondrial dysfunction caused by application of 3-nitropropionic acid, cellular degeneration in the cochlear lateral wall primarily accounts for hearing impairment due to acute energy failure [68]. These clinical entities could be candidate diseases for regenerative medicine in the cochlear lateral wall, whereas much works should be done to elucidate the pathophysiology of each clinical condition in the future.
5.6 Conclusion
In this chapter, we presented a brief review on the cochlear lateral wall from the viewpoint of regenerative medicine. Recent progress in the field of research in the cochlear lateral wall in the last two decades has made our knowledge on the lateral wall biology remarkably improved. At the same time, we recognize that we are still at an early stage in our understanding of the molecular biology and physiology of the cochlear lateral wall. It would depend on further studies in the future to unveil the precise roles and mechanisms of the lateral wall under normal and affected conditions and develop translational researches to connect basic biology to regenerative medicine.



