
FIGURE 6.1 A. Type 1 CNVM. Left. FA showing leakage of undetermined origin. Right. ICG angiography showing arteriolized pattern of choroidal neovascularization. B.Type 1 CNVM. Left. ICG angiography showing arteriolized pattern of CNV. Right. Macular OCT showing a visible large pigment epithelium detachment (PED).
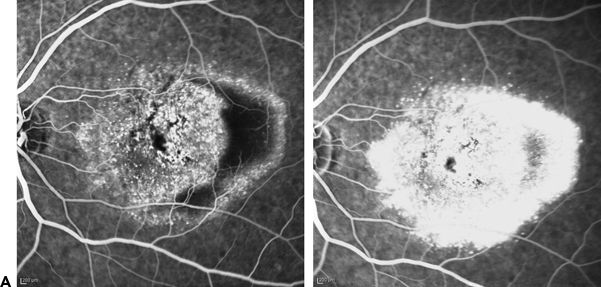

FIGURE 6.2 A. Type 1 CNVM. Early- and late-phase FA: Leakage of undetermined origin. PED is visible. B.Type 1 CNVM. Left. ICG: Bushy appearance of CNVM apparent with ICG. Right. OCT of the same case. Pigment epithelium detachment with minimal subretinal fluid.
Spectral-domain optical coherence tomography (SD- OCT) allows visualization of intraretinal structures as well as RPE morphology (Figs. 6.1B and 6.2B). It identifies RPE detachment and CNV-related infiltration. According to Malamos et al. (14), with the use of the RPE map, it is possible to identify the location of the occult CNVM. If a membrane is hidden under the RPE, the latter appears slightly prominent, flat at the top, and slowly declining at the margins. These lesions are usually ill defined and convex with irregular surfaces. SD-OCT can be more informative than is FA and allows better identification and classification of membranes with occult components that may not be easily detected and delineated. The 3D surface maps of the RPE further clarify the exact level of the lesion and its microscopic morphology in combination with B-scan morphology.
Type 1 membranes are the most common type associated with AMD, while type 2 membranes are associated with focal lesions affecting the Bruch’s membrane and the RPE in diseases such as high myopia with lacquer cracks, ocular histoplasmosis syndrome, serpiginous choroiditis, and choroidal ruptures (20). In disorders like AMD, lipid-rich material, such as cuticular drusen, contains numerous complement components that accumulate between the Bruch’s membrane and the RPE. Type 1 neovascularization grows in the presence of such material and is not seen in disorders with focal disturbances of the RPE–Bruch’s membrane complex such as multifocal choroiditis and panuveitis, myopia, and choroidal rupture (10). Type 1 pattern is rarely seen in disorders like Best disease, vitelliform macular dystrophy, and pseudoxanthoma elasticum where material accumulates above and below the RPE monolayer (10).
Unlike classic lesions, occult-type lesions rarely cause visual symptoms promptly, allowing some lesions to grow to a large size before being detected clinically. They tend to have a variable and less aggressive natural course based on the presenting acuities and long-term natural history. In the MARINA study (21), patients with occult neovascular lesions presented better initial visual acuities than in the ANCHOR study (22) patients who had classic lesions. This was also observed in the VIP and TAP trials where many minimally classic lesions and occult lesions with no classic component had relatively acceptable visual acuity (23). According to Grossniklaus et al. (24), this benign course may relate to the theory that in some eyes, type 1 neovascularization may grow as a compensatory mechanism to provide nutritional support to the ischemic outer retina by recapitulating the normal choriocapillaris. In his study, Engelbert et al. (25) observed that only one eye out of eighteen eyes with type 1 neovascularization (6%) developed geographic atrophy overlying affected areas.
The exudation in type 1 neovascularization is predominantly in the form of subretinal fluid. Subretinal fluid is less clinically aggressive than is cystoid macular edema (CME). The presence of CME portends a poor prognosis, in that it presumably indicates damage to the outer retinal barrier in the form of RPE dysfunction and disruptions in the tight junctions that contribute to the external limiting membrane (ELM) band on SD-OCT (3). However, in some cases, type 1 neovascularization may follow an aggressive course and cause a decrease in visual acuity. These vessels may also erode through the RPE giving rise to type 2 neovascularization. In the TAP study, approximately 40% of eyes progressed from minimally classic to predominantly classic lesions with 24% converting in the first 3 months (26). This was also confirmed by Schneider et al. (27) when 23% of eyes with occult lesions having no classic components progressed to predominantly classic membranes over a period of 6 to 12 months. Overall, the conversion rate from occult with no classic to classic neovascularization at 1 year was 46% (39%–54%); no conversion in the reverse direction was ever described (28).
The response to treatment with intravitreal anti–vascular endothelial growth factor (VEGF) therapy may be less than the response observed in type 2 or type 3 neovascularization. Eyes with these type of neovessels may often continue to manifest extrafoveal subretinal fluid. Type 1 neovascularization may be less responsive to anti-VEGF treatment; nevertheless, the antipermeability effects of these drugs are often sufficient to resolve most, if not all, of the fluid above the RPE monolayer and may limit the continued growth of these vessels. Despite the incomplete clearance of subretinal fluid, and sometimes the CME, many eyes maintain stable visual acuity for up to 5 years on monthly anti-VEGF treatment with no eyes experiencing sight-threatening submacular hemorrhages (25).
It was described by Spaide (29) that vessels may be detected by SD-OCT as hyperreflective material lining the undersurface of the elevated RPE. The contraction of this neovascular tissue, spontaneously or in response to intravitreal anti-VEGF treatment, could produce dehiscence and tears in the RPE layer. Such possibility can be predicted by the linear diameter of the PED as well as the maximal PED height (30).
TYPE 2 NEOVASCULARIZATION
As mentioned previously, the type 2 pattern predominates in macular disorders where there is a focal alteration of the RPE–Bruch’s membrane complex such as pathologic myopia with lacquer cracks (Fig. 6.3A and B), punctate inner choroidopathy, multifocal choroiditis and panuveitis, histoplasmosis, and choroidal rupture. It is seen also in entities where material is deposited above the RPE such as vitelliform macular dystrophies, Best’s disease, Stargardt’s disease, subretinal drusenoid deposits (reticular pseudodrusen), and pseudoxanthoma elasticum (10).


FIGURE 6.3 A. Type 2 membrane in a case with high myopia. FA (early and late phases) showing leak in a classic pattern. B. OCT of the same case. Disruption of the RPE is observed as well as a hyperreflective area anterior to the RPE that corresponds to a type 2 CNVM.
Type 2 lesions usually start as type 1 neovascularization and then can penetrate the RPE–Bruch’s membrane complex to proliferate in the subretinal space above the RPE monolayer to give the type 2 appearance (28).
Histopathology studies show that predominantly classic neovascularization is mainly subretinal or combined subretinal with sub-RPE components. Classic membranes were seen to contain not just capillaries but also larger caliber vessels, whereas occult membranes contained predominantly, or only, capillaries (12).
According to MPS criteria, CNV was considered well defined or classic when a net of new vessels was identified at an early stage of FA with progressive leakage in later stages (Fig. 6.4A and B). They fluoresce intensively against a relatively dark background due to attenuation of the choroidal fluorescence by the intervening RPE (Fig. 6.5A). About 17% (11) of all CNVM show exclusively classic membranes, but it is possible that many of these harbor small areas of type 1 vessels that are undetectable by FA alone (10). Occult lesions are sometimes underestimated due to overlying classic membranes, but a PED seen on OCT can explain the presence of an occult membrane. Classic membranes can also be seen with ICG as an area of hyperfluorescence without marked leakage activity. ICG images may underestimate the dimension of the neovascular complex compared to morphologic imaging by SD-OCT (31).
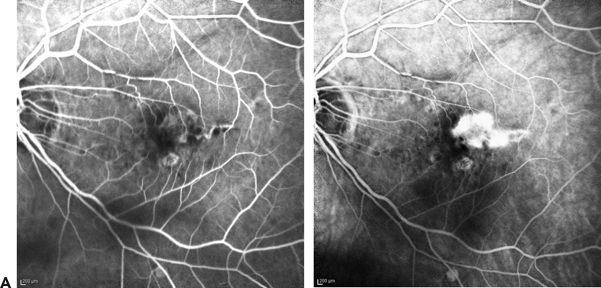

FIGURE 6.4 A. FA showing CNVM (early and late phase) with leakage in a classic pattern. B. OCT of the same case: Intact RPE with an overlying hyperreflective lesion representing a type 2 CNVM.


FIGURE 6.5 A. FA showing CNVM (early and late phases) with leakage in a classic pattern. B. OCT of the same eye showing disruption of the RPE with an overlying hyperreflective lesion associated with subretinal fluid.
On OCT, a high level of correlation was found between areas of classic CNV seen on FA and the volume of subretinal tissue (32). It is possible to differentiate a classic CNV using SD-OCT. The retinal thickness map specifically shows a well-defined lesion with steep borders and a craterlike central depression (14). Leakage and staining can be easily differentiated with this imaging technique where hyperreflectivity represents visualization of fibrotic tissue and not edema. With OCT, type 2 membranes can be localized above the RPE and beneath the photoreceptor outer segments (10) (Fig. 6.5B).
The hyperreflective line, anterior to the RPE line, represents the inner segment–outer segment junction of the photoreceptors (33). Disorganization of this layer, overlying the inner segment–outer segment (IS/OS) junction, is often accompanied by intraretinal cystic spaces. Intraretinal fluid rather than subretinal fluid predominates in type 2 lesions and usually is associated with decreased visual acuity (34,35). The differentiation between these two types of leakage can be easily seen with OCT but is not possible with FA (10).
In patients with classic lesions, most vision loss occurs within the first 6 months with little vision loss after the first year. Predominantly classic lesions included in the TAP trial were found to be smaller in size than the occult (type 1) lesions included in the VIP trial. It is suggested that predominantly classic lesions almost always cause visual symptoms promptly, perhaps allowing identification of smaller lesion sizes, whereas occult lesions may or may not cause prompt visual symptoms, which allows them to grow to larger sizes (23,28).
Early in their evolution, type 2 vessels appear to be exquisitely sensitive to intravitreal anti-VEGF agents. There may be a decrease in the volume of subretinal fluid and fibrovascular tissue. In larger and more mature lesions, the abnormal vessels remain as a hyperreflective band (subretinal fibrosis) causing disorganization and thinning of the overlying receptors. This is seen as a disruption of the IS/OS junction, loss of the ELM, and thinning of the outer nuclear layer on SD-OCT. These eyes may continue to manifest intraretinal fluid despite frequent treatment, presumably due to the loss of the outer blood–retinal barrier in the form of RPE damage and disruption of the tight junctions that contribute to ELM band on the OCT (10). Even in eyes with early lesions, the disruption of the IS/OS layer may be indicative of the future visual result, and visual recovery may be limited in these eyes because of the initial assault to the photoreceptors (33).
TYPE 3: RETINAL ANGIOMATOUS PROLIFERATION
It was a long-held belief that all neovascularizations in AMD originated from the choroidal circulation. In 2001, Yannuzzi et al. (36) challenged this traditional dogma based on the presumed origin and evolution of the neovascularization process. They believed that IRN originating from the inner retinal circulation produced a surrounding telangiectatic compensatory response to accommodate an increase in vascular perfusion, and they termed this variant “retinal angiomatous proliferation” (RAP). Thereafter, and due to a lingering uncertainty as to the origin of these lesions, Freund et al. (37) have proposed naming this vascular lesion “type 3 neovascularization” to distinguish and emphasize the location of the neovascularization (intraretinal) independent of its origin. Type 3 neovascularization, or RAP, indicates proliferating vessels within and below the retina itself. The origin of this neovascular subtype can be from either circulation and may originate from both circulations simultaneously in the form of a retinal–choroidal anastomosis (RCA). The presence of an RCA gives this entity many of its characteristic clinical, SD-OCT, and angiographic features including intraretinal hemorrhages and CME. Type 3 lesions have been observed originating in areas with substantial photoreceptors, bringing the deep retinal vasculature in close proximity to the underlying RPE, Bruch’s membrane, and choriocapillaries. Typically, the angiogenic response occurs overlying a focal drusen/basal laminar deposit that may already be infiltrated by type 1 vessels. Presumably, loss of or erosion through the intervening RPE and an appropriate angiogenic milieu would allow the retinal and choroidal circulations to merge in the form of RCA with a continued proliferative response. As these lesions originate from both the retinal and choroidal circulations, they never originate within the foveal avascular zone.
RAP is further subdivided into three stages, which describe the disease progression. Stage I indicates the earliest clinical manifestations, in which RAP begins in the deep retinal complex forming IRN. As the vessels evolve, they extend beneath the neurosensory retina, becoming subretinal neovascularization (SRN), or stage II. In stage III, an RCA is formed as SRN merges with choroidal circulation, forming CNV (38–41) and the potential for a vascularized pigment epithelium detachment (PED). FA is useful in revealing the presence of the angiomatous intraretinal vascular complex and the extension of the associated PED. However, other diagnostic techniques, such as indocyanine green (ICG) angiography and OCT, have now proven to be as useful—or even more—to demonstrate the presence of the RAP lesion.
Treating RAP has proven difficult due to the unique nature of the disease and a lack of clinical trials focused on this form of neovascularization. To date, only prospective and observational studies have been conducted, often with small sample sizes, and no standard of treatment has been established. As with other forms of neovascularization, the main treatment options for RAP include anti-VEGF agents, PDT, focal laser, transpupillary thermotherapy, and surgical ablation, or various combinations of these. To date, PDT with intravitreal triamcinolone provided the most promising results initially, but by 12 months, many of the effects had faded (42). Anti-VEGF agents may be beneficial in decreasing vasogenic drive and retinal edema, when followed by PDT (43). ICG-guided focal laser obliteration as salvage treatment may be an option, as proposed by Bearelly et al. (44).
TYPE 4: POLYPOIDAL CHOROIDAL VASCULARIZATION
Polypoidal choroidal vascularization (PCV) is recognized as a subset of type 1 CNV due to the location of PCV abnormalities, below the RPE. The various clinical manifestations of PCV are shown in Figure 6.6. PCV is most frequently characterized by a branching network of choroidal vessels and orange-red polypoidal lesions (45). PCV vascular abnormalities lie between the RPE and Bruch’s membrane (46); therefore, PED is often seen in patients with PCV (47,48). PCV has often been misdiagnosed as other forms of exudative AMD due to similarities in clinical appearance on OCT and FA (49). Both PCV and CNV cause hemorrhages and other signs of exudation, leading to scarring and fibrosis, and have similar genetic markers; however, PCV has significant differences from CNV in epidemiology, pathophysiology, clinical course, and treatment response.

Figure 6.6 Common presentations of PCV, including branching vascular network, PED, exudation, and subretinal hemorrhage. Illustration by Mariana Ingolotti.
When PCV was first described, it was thought rare and was once recognized as a disease that primarily affected black women (50). It is now known that PCV affects people of all races, although at varying rates. PCV diagnoses are given most frequently within Asian populations (51), where PCV is thought to be as common as wet AMD is in Caucasian populations (48,52,53). Although they are less affected by PCV, two studies focusing on Caucasian patients indicate that PCV is not specific to “pigmented races” (54,55) and that Caucasian patients, especially those with extramacular lesions, should be examined for PCV (53,54). The age of diagnosis for PCV is typically younger than that for exudative AMD, between the ages of 60 and 70 years (56).
PCV, with its varying presentations and response to treatment, is in need of its own classification system. Several possible schemes have been proposed (Table 6.1). The first scheme divides PCV into three groups, relating it to CNV. In group one, the most common type, choroidal vascular abnormalities are observed beneath the RPE. In group two, PCV and CNV are present, expanding beneath the RPE with polypoidal lesions developing at vessel termini. And, in group three, CNV develops secondary to radiation treatment (60). A second scheme is based on polyp size, location, formation, and number of polyps (57). The third scheme divides the disease into exudative and hemorrhagic groups. The exudative group is characterized by a serous PED associated with intraretinal lipid deposits in the macula. It mimics chronic central serous chorioretinopathy in the elderly. The hemorrhagic group is characterized by hemorrhagic PED and subretinal hemorrhage, mimicking exudative AMD (58). All of these schemes are designed to better explain the clinical course of the disorder and tailor treatment options to individuals.
Table 6.1 PROPOSED SCHEMES FOR THE SUBCLASSIFICATION OF PCV

Genetic research may eventually lead to a more objective classification scheme for PCV and the other forms of macular degeneration. Genetic studies have found that PCV and AMD have similar genotypes and major alleles in both Japanese and Caucasian patients. The major loci associated with AMD and their haplotype-tagging alleles (CFH, CFB/C2, and ARMS2/HTRA1 with rs547154, rs1061170, rs1410996, and rs10490924) were also significantly associated with PCV (59,61–63). While genetic studies demonstrate similarities between the two disorders, many differences are seen in tissue studies.
Examinations of PCV tissue have been small in number but provide many clues into the pathogenesis of PCV and its relationship to CNV. Most histology studies have focused on vasculature, specifically the branching vascular networks unique to PCV. These analyses have been unable to determine the pathogenesis of this characteristic. Two theories seem plausible: (a) The vessels stem from new CNV, and (b) the vessels are existing choroidal vessels that have dilated abnormally. Histologic findings show that vascular endothelial cells within PCV specimens were negative for VEGF (48,64). As VEGF is primary to CNV development, its absence may suggest that the dilated vessels stem from existing choroidal vessels (64).
Vessel pathology shows large choroidal arterioles and venules, with the arterioles having an inner elastic layer, a feature not present in CNV. These arterioles are located below the Bruch’s membrane and may explain the pulsation often observed in ICG angiography (65), a characteristic of PCV that distinguishes it from CNV (46,66,67). Pulsation may represent an abnormality of inner choroidal vasculature, increased intravascular pressure, and greater physical stress on the vessel wall (55). Pulsatile lesions have a different pathophysiologic mechanism and, therefore, may respond differently to treatment (68). Another distinct vascular characteristic of PCV is the hyalinization of choroidal vessels with arteriosclerotic changes (Fig. 6.7A and B). This is unique to PCV, as hyalinization has not been reported in eyes with CNV (69).


FIGURE 6.7 A. Photomicrograph showing dilated vessels (asterisk) beneath the RPE (arrowheads) that were thickened and hyalinized. (Lafaut BA, Aisenbrey S, Broecke C, et al. Polypoidal choroidal vasculopathy pattern in age-related macular degeneration. A clinicopathologic correlation. Retina. 2000;20:650–654; Nakashizuka H, Mitsumata M, Okisaka S, et al. Clinicopathologic findings in polypoidal choroidal vasculopathy. IOVS. 2008;49(11):4729–4726.) B. Illustration of dilated and hyalinized vessels, shown in (A), as they are located between the Bruch’s membrane and the RPE. Illustration by Mariana Ingolotti.
Other histologic findings link PCV with CNV. In some PCV eyes, fibrovascular membranes have been located within the Bruch’s membrane (70,71). In one case, clusters of thin-walled dilated vessels correlated with polypoidal structures on ophthalmoscopy and ICG angiography, indicating that the fibrovascular complex was subretinal CNV. However, this eye had previously undergone radiation for a subretinal neovascular membrane, making radiation associated choroidal neovasculopathy a possibility (72).
Nakashizuka et al. (69) found that PCV and CNV can be found simultaneously in the same eye, either by chance or, as they speculate, secondary to a wound repair reaction. Massive exudation of fibrin and blood plasma from dilated hyalinized choroidal vessels may raise choroidal tissue pressure, leading to protrusion of choroidal tissues through the RPE and Bruch’s membrane. These cases resulted in a classic CNV pattern on FA.
Histologic studies, OCT, FA, and ICG angiography have all demonstrated distinctions between PCV and other forms of exudative AMD. Many of these studies have led researchers to conclude that PCV is caused by inner choroidal abnormalities, not CNV (1,40,51,60,73).
In 1999, OCT was first used to examine PCV. Iijima et al. (74) showed that cross-sectional OCT images of the retina accurately located polypoidal structures seen on ICG angiograms. Since then, other studies have proven that choroidal hyperreflectivity on OCT tomograms can accurately locate the choroidal abnormalities demonstrated on ICG and aid in the diagnosis of PCV (75,76), as shown in Figure 6.8. In particular, PED, branching vascular networks, and polypoidal lesions can be located (78,79). SD-OCT reveals the Bruch’s membrane as a line of high reflectivity beneath vascular abnormalities caused by PCV (80). For some patients with PCV, a double-layer sign, consisting of the RPE and another hyperreflective layer, is seen on OCT in the area of network vessels, particularly in eyes with serous retinal detachment (81). OCT scans have been used to compare the macular and choroidal thickness in PCV eyes to those with other forms of exudative AMD. Koizumi et al. (82) used spectral-domain OCT and found thickening in the choroid of PCV eyes. When subfoveal choroidal thickness measured greater than 300 μm, patients were 5.6 times more likely to have PCV. These results suggest that there may be significant structural differences in the PCV choroid as compared to typical exudative AMD, most likely stemming from dilation of choroidal vessels or choroidal vascular hyperpermeability as seen on ICG. Enhanced depth imaging OCT also demonstrated choroidal thickening in patients with PCV and showed that in exudative AMD, choroidal thinning was more likely to occur (83).



Figure 6.8 PCV in a 75-year-old man. A. Funduscopic examination shows two reddish orange nodules (arrows) with serous PED. B. ICG shows branching vascular network (arrowhead) terminating in polypoidal lesions (arrows). Temporal polypoidal lesion accompanies serous PED. OCT images. C. show branching vascular network (arrowheads) terminating in polypoidal lesion (arrows). (Tsujikawa A, Sasahara M, Otani A, Gotoh N, et al. Pigment epithelial detachment in polypoidal choroidal vasculopathy. Am J Ophthalmol. 2007;143: 102–111.) (Ref. 77).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


