Chronic Iridocyclitis
William A. Godfrey
The patient with chronic iridocyclitis and anterior segment inflammation is a difficult problem from the standpoint of both diagnosis and management. The options for anti-inflammatory management become more difficult as the clinician tries to manage the risks of long-term local corticosteroid side effects and use of other local anti-inflammatory agents balanced against systemic side effects of longer-term use of oral or systemically administered anti-inflammatory agents. In addition, even if the diagnosis is known, treatments may often be ineffective.
In chronic iridocyclitis, the iris, and ciliary body are the primary sites of active chronic or long-term inflammatory processes, which last for more than 3 months. Other tissues, such as the cornea, sclera, pars plana, and peripheral choroid and retina may be involved in anterior segment inflammation and may secondarily involve the iris and ciliary body because of their close proximity to these chronically inflamed nearby structures. Chronic iridocyclitis characteristically has an insidious onset and may sometimes be asymptomatic, so that it may be present for some time before it is recognized clinically. The activity of an acute iridocyclitis may decrease, progressing to chronic iridocyclitis. Chronic iridocyclitis may be a persistent inflammation lasting months or years, or it may consist of recurrent, active episodes with periods of minimal or no inflammatory activity interspaced in between. Recurrences tend to be similar in clinical course to previous episodes. There may also be significant, constant activity and episodes of increased inflammation. Chronic inflammation may be due to tissue damage and subsequently altered structure, rather than to active chronic inflammatory processes.
Chronic iridocyclitis is frequently noted coincidentally or because of decreased vision. There is usually little pain. Photophobia and the other symptoms of inflammation are usually absent or minimal. There may be many of the findings seen in acute iridocyclitis, including cells, flare, keratic precipitates, anterior and posterior synechiae, secondary cataract changes, and secondary glaucoma. Band keratopathy, iris nodules, vitreal debris, and macular edema are more characteristically associated with chronic iridocyclitis. These signs may be quantitated and characterized to serve as indicators of inflammatory activity for evaluating levels of activity and effectiveness of management. The characteristics of these findings are useful in the clinical diagnosis of certain conditions.1 Standardization of this terminology used in evaluating and recording these findings and characterizations has been updated and solidified by the Standardization of Uveitis Nomenclature working group (SUN).2
These processes tend to be self-limiting, but the damage done by chronic inflammation can be very destructive to eye tissues, especially if activity is present for months or years. The physical property of ocular transparency, as well as the passage to and focusing of a sharp image on a receptive and functioning retina, is unique to the eye as a necessary physiologic process, and impairment of this property is a defining characteristic of pathologic process. Ocular transparency is very vulnerable, and systemic disease processes that are minor in terms of their effect on other body processes can have a disastrous effect on this physical property. Diagnostic tests that are used to detect levels of systemic disease producing widespread tissue injury frequently do not have the specificity or sensitivity to detect low levels of injury and are disregarded or are considered negative when they are not at levels expected in systemic widespread disease. Conventional laboratory testing frequently does not recognize the lower levels of systemic inflammatory disease that also involves the eye and will produce damaging ocular inflammatory injury. The situation is frequently encountered where a specific etiology or mechanism is not identified, or may be only suspected. When a specific treatment is not available, the management is directed at minimizing the damage from the inflammation so that the eye will be capable of the best salvageable function when the inflammation resolves. In chronic processes, the medications used to suppress the inflammatory process can sometimes be more damaging than the disease process itself. An understanding of the disease process and its effect on the eye must be carefully weighed against the long-term effects of medications used in treatment.
Iridocyclitis is basically an anatomic diagnosis, where the iris and ciliary body are the primary areas of inflammatory involvement. As in acute iridocyclitis, signs of chronic iridocyclitis can be present as a spillover from anterior inflammation (e.g., keratitis, primary scleritis), from posterior segment disease (e.g., Toxoplasma retinochoroiditis), or from noninflammatory masquerade syndromes. These other conditions must be considered in the diagnosis of chronic anterior segment inflammatory disease and are covered in other chapters. Multiple causes can produce similar clinical pictures with only subtle differences in the character of the findings. A careful history, review of systems and appropriate laboratory tests, specialty consultations, and repeated observations may be necessary to help clarify the nature of a chronic inflammatory process.
JUVENILE RHEUMATOID ARTHRITIS (JRA)
Chronic Arthritis in Children
The terms juvenile rheumatoid arthritis (JRA), juvenile idiopathic arthritis (JIA), and juvenile chronic arthritis (JCA) are not interchangeable and are based on the definitions established by the American College of Rheumatology (for JRA), the European League Against Rheumatism (for JCA) and the Pediatric Task Force of the International League of Associations for Rheumatology (ILAR) (for JIA), all attempting to make the diverse clinical presentations fit into a standard classification. The comparison of definitions of JRA and JIA is shown in Table 1, and the ongoing refining of this classification by the ILAR is expected. Historically the older classification of the American College of Rheumatology has been the classification for previously published literature and will be used here for this discussion. A suggested compromise for this has been to use the terms juvenile arthritis or chronic arthritis in children, regardless of which classification is used.
TABLE 1. Comparison of Classifications for Juvenile Rheumatoid Arthritid and Juvenile Idiopathic Arthritis | |||
|---|---|---|---|
|
Juvenile arthritis or chronic arthritis in children seems to represent a group of diseases with similar clinical characteristics and multiple causes.3 For present practical discussion of these differences, the older JRA classification used in the older basic literature seems best. JRA has been divided by the American College of Rheumatology into clinical subsets that attempt to group clinical disease into three types:
Polyarticular JRA is the most common form of the disease (30%). Joint involvement is the major symptom. It is characterized by involvement of five or more joints during the first 6 months of the disease, with a combination of pain, swelling, and limitation of movement. Iridocyclitis develops in approximately 5% of these patients.
Systemic or Still’s disease, the more severe systemic form of JRA, accounts for 10% of JRA and is associated with fever, leukocytosis, rash, myalgia, splenomegaly, lymphadenopathy, pleuritis, and hepatomegaly. It does not always have joint involvement initially but frequently goes on to become polyarticular disease. This type is rarely associated with iridocyclitis.
Oligoarticular and pauciarticular JRA account for 60% of JRA and have two groups that are associated with both immunologic markers and iridocyclitis. One group (A1) is HLA-B27–positive patients who tend to have episodes of acute iridocyclitis similar to those seen in the spondyloarthropathies, tend to go on to develop sacroiliitis, and probably represent juvenile onset of these diseases. The other group (A2) is antinuclear antibody (ANA) positive and includes patients who tend to develop the chronic iridocyclitis more commonly associated with JRA.3,4,5,6 A2 is associated with early onset oligoarthritis disease in girls. HLA-B27 seems related to juvenile onset of spondyloarthropathy, but there is some indication of age-related risk for oligoarthritis. There appears to be a genetically associated predisposition. Certain DR, DQ, and DP alleles seem significantly associated with chronic pauciarticular JRA, but not with the appearance of iridocyclitis, whereas DRB1*01 may seem to have a protective effect against the development of iridocyclitis (DRB1*1301, DRB1*801, DRB1*1104, DQA1*0401, DQB1*0402, DPB1*0201).7,8,9,10
In ANA-positive pauciarticular JRA patients, the age at onset ranges from 6 months to 15 years, but it has occurred in adulthood.11 The highest incidence of onset is from 1 to 4 years of age. Systemic symptoms are generally milder. Joint involvement is usually insidious. Weight-bearing joints, particularly the knees, are most commonly involved. This involvement may be asymmetric. An increased incidence of women is seen in the ANA-positive group with iridocyclitis, and an increased incidence of men is seen in the HLA-B27–positive group.6
Arthritis must persist for at least 6 weeks in one to four joints, when other subtypes are excluded. Other diseases must be excluded, as well, including infectious arthritis due to viral, bacterial, tuberculous, or fungal etiologies. Ankylosing spondylitis, Reiter’s syndrome, systemic lupus erythematosus, rheumatic fever, vasculitis, orthopedic disorders, and many other conditions can present in this manner. A rheumatologist may be necessary to clarify the diagnosis, and synovial fluid analysis and biopsy may be needed.6,12
The pauciarticular group has been reported to account for 75% to 95% of all iridocyclitis associated with JRA. The arthritis usually manifests before iridocyclitis is noted. Reports suggest arthritis may be present 1 to 10 years before iridocyclitis; however, iridocyclitis may be the presenting disease.
The HLA-B27–positive group tends to have episodes of acute iridocyclitis with redness, pain, and light sensitivity lasting 3 to 6 weeks and follows the course of the adult iridocyclitis seen with adult spondyloarthropathies.
In the ANA-positive group, the iridocyclitis is usually asymptomatic and insidious in onset. Girls are affected more frequently than boys, with a ratio of about 4:1. Light sensitivity, mild discomfort, redness, tearing, or spots in vision may be the only symptoms. Even when aware of these symptoms, children usually do not complain about them. Evidence of iridocyclitis may be found without symptoms during a routine examination or when the patient has been referred for a periodic examination by the pediatrician, rheumatologist, or other physician aware of this frequent complication association with pauciarticular JRA. A cataract or irregular pupil due to posterior synechiae (Figs. 1 and 2) may be the first sign of ocular involvement.13,14,15
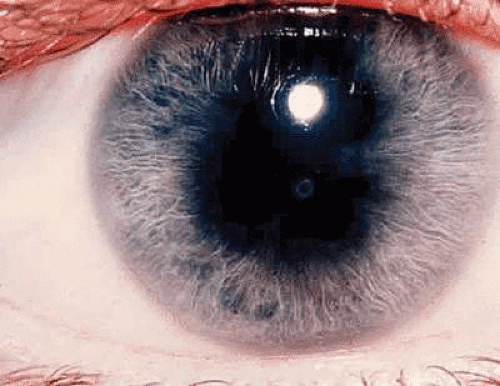 Fig. 1. Multiple posterior synechiae in juvenile rheumatoid arthritis. |
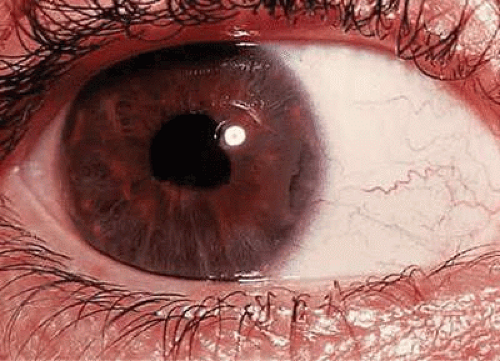 Fig. 2. Extensive posterior synechiae formation with band keratopathy in juvenile rheumatoid arthritis. |
There seems to be little correlation between the activity of the joint inflammation and the iridocyclitis. Bilateral involvement has been reported in up to 50% of patients. Band keratopathy (13% to 49%) (Fig. 2), cataract (22% to 58%), and posterior synechiae (38% to 61%) are frequent complications. Glaucoma is also a serious complication and has been reported in 14% to 22%.13,16 Vitreal debris, disc edema, and macular edema do occur but are rarely associated with long-term morbidity. The posterior pole is not usually involved except when there is secondary spillover of severe anterior segment reaction into the vitreous.14,17,18,19,20,21
When cataracts are severe enough to require surgical removal, newer extracapsular surgical procedures and the use of additional local or systemic corticosteroids at the time of surgery have been reported to improve results. Posterior pars plana lensectomy and removal of any cyclitic membranes present have also improved the prognosis when this complication is involved. Earlier series reported a poor prognosis following surgery, with 48% having a final visual acuity of 20/200 (6/60, metric equivalent) or less.22 More recent series with improved techniques suggest a better prognosis.23 Pretreatment and extended postsurgical treatment with topical and oral corticosteroids seem to help control associated inflammatory activity. Surgical procedures for cataract removal are best delayed until the inflammatory process has been inactive for 6 to 12 months. Intraocular lens implants are still at risk for serious complications; with chronic inflammation, displacement of the lens is a frequently noted complication, along with pigment and cell precipitates on the lens, synechiae of the iris to the lens, increased activity of the inflammation, secondary glaucoma, and vitreoretinal complications.23 Intraocular lens implantation in these circumstances should be delayed.
The chronic persistence of inflammation makes this iridocyclitis particularly difficult to control. Cycloplegia and topical corticosteroids are sometimes sufficient in mild disease and may be necessary on a long-term basis. Constant reevaluation and adjustment of treatment is necessary in active disease. Oral corticosteroids may be needed in severe disease. The long-term use of oral corticosteroids can produce severe side effects, especially in children, and their use must be seriously weighed in terms of therapeutic benefits versus disability produced by the treatment. Systemic nonsteroidal anti-inflammatory agents, such as tolmetin and naproxen, seem to have a significant place in the treatment of childhood intraocular inflammation, if they are well tolerated. They must be used on a longer-term basis, however, and response to treatment takes months to assess. The currently understood side effects, however, make their long-term use appear safer than alternative long-term treatments.3,24 Naproxen has been noted to cause a condition called pseudoporphyria, and was associated with significant facial scarring in 12% in one study.25,26,27 Renal side effects, including glomerular microproteinuria, have been reported in association with the use of nonsteroidal anti-inflammatory agents in JRA treatment.28 The addition of biologicals such as etanercept, infliximab adalimumab, and others seem to have significant promise, but larger experience is needed to evaluate their long-term usefulness and side effects. The use of methotrexate has increased significantly, and mycophenolate mofetil may have a place in more aggressive therapy but needs more evaluation. These treatments are best evaluated and directed by the pediatric rheumatologist.
The presence of antinuclear antibodies in patients with JRA allows the separation of a group particularly prone to the development of iridocyclitis. An 88% incidence of positive antinuclear antibodies in patients with iridocyclitis suggests that this group has a significantly increased risk of iridocyclitis.14 These patients should be examined regularly with the slit lamp for signs of iridocyclitis. When iridocyclitis has been present, but in remission, slit lamp examinations are suggested every 1 to 3 months. When there are no signs of inflammatory activity, a slit lamp examination approximately every 3 months is appropriate. Children with polyarticular JRA who are ANA negative and have no ocular inflammation should be examined at least yearly.16 Adult-onset JRA is occasionally seen, and the extension of active disease into adulthood is also noted in the pauciarticular group with eye involvement.29
Chronic Iridocyclitis in Young Girls
A clinical condition of chronic iridocyclitis in children (particularly young girls) has been described that is very similar to the iridocyclitis seen in pauciarticular JRA, but there is no joint involvement. The female-to-male incidence is reported to be 12:1. ANA positivity has been noted in several of these patients tested because of the recognition of the value of this test in pauciarticular JRA. These patients probably represent JRA patients without arthritis who may become arthritic later if the localizing precipitating factors of the joint manifestations occur. It is recognized that iridocyclitis readily occurs in monarticular and pauciarticular joint disease, so it is reasonable to expect that in a few of these patients the eye disease will occur, but the joint disease will be so minor that it either goes unrecognized or may not occur at all.30,31
HETEROCHROMIC IRIDOCYCLITIS
Fuchs’ heterochromic iridocyclitis is characterized as a unilateral chronic iridocyclitis of insidious onset. Small, white, round keratic precipitates (Fig. 3) usually are scattered uniformly over the superior and inferior cornea, with fibrin stellate-shaped flecks on the endothelium. Cells and flare are present in the anterior chamber but are sometimes minimal. Thinning of the iris stroma (Figs. 4 and 5) leads to the change in iris color. Iris transillumination is sometimes visible in the affected iris owing to loss of stroma and pigment epithelium of the iris. It is rarely bilateral, so that the ability to recognize the contrast in iris stroma changes, as well as the distribution of the characteristic corneal precipitates, may help to make this diagnosis. Posterior subcapsular cataract changes (Fig. 6) may progress to a maturing cataract (Fig. 7) in the later stages. Because the disease process may be asymptomatic, the cataract changes may produce the initial symptoms of decreased vision. Posterior subcapsular changes are similar to those seen in other chronic iridocyclitis and are probably due to the metabolic effects on the lens from changes in the aqueous. Vitreous cells and coarse opacities are usually present, and no posterior pole abnormalities are usually observed.32,33,34,35 Reports of heterochromic iridocyclitis associated with inactive Toxoplasma retinochoroiditis and sarcoidosis suggest a correlation may exist, at least in some clinical situations.36,37,38,39,40 This indicates that there may be a common ultimate clinical response in the eye that is related to several causes.
 Fig. 3. Small, white, round keratic precipitates in Fuch heterochromic iridocyclitis. |
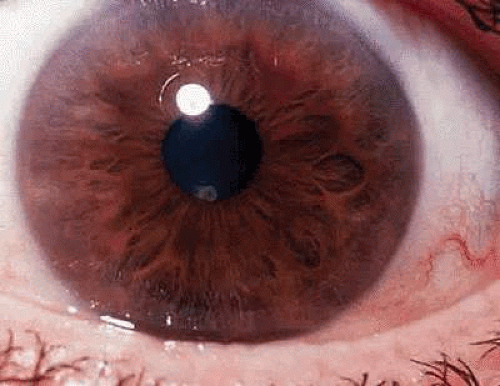 Fig. 4. Normal iris of patient with heterochromic cyclitis. |
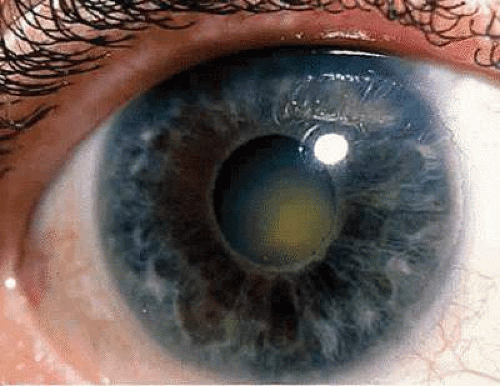 Fig. 5. Involved eye showing iris stromal thinning, peripapillary iris pigment epithelial loss, and alteration in color compared with the normal eye (Fig. 4). |
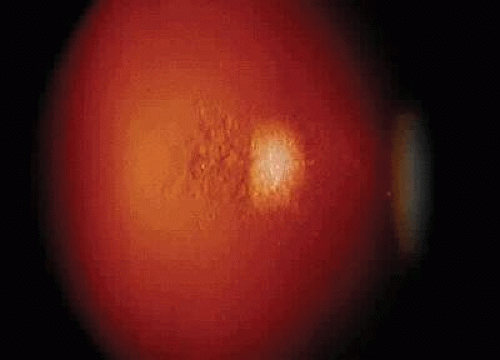 Fig. 6. Posterior subcapsular cataract changes in heterochromic cyclitis. |
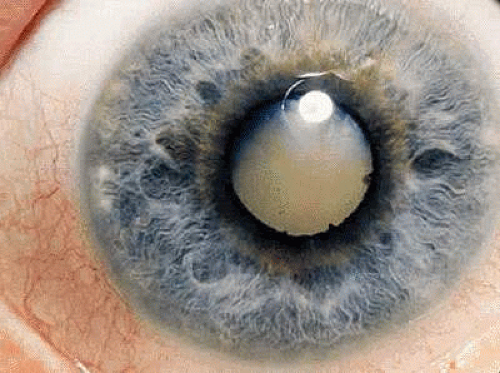 Fig. 7. Maturing cataract changes in heterochromic cyclitis. |
Heterochromic iridocyclitis is a frequently undiagnosed condition, especially in brown-eyed persons in whom the brown pigment may obscure the iris stromal changes. Loss of the sharpness of detail in the iris rugae is observed.41,42 The loss of stromal substance and thinning of the iris is usually diffuse, involving the entire iris, and does not involve sectors as in herpes simplex uveitis or herpes zoster uveitis. A diffuse herpes simplex or zoster iridocyclitis could produce diffuse stromal atrophy, and increased incidence of herpes simplex DNA has been reported in one series of patients where the aqueous was studied. Additional series report increased rubella antibodies production.43,44,45 Appearances may resemble iris atrophy seen in other conditions. This is usually easily differentiated by the somewhat unique distribution of 360° over the endothelium of the keratic precipitates and their appearance, along with the fibrin stellate flecks between precipitates. Its frequency has been reported to be as high as 2% to 3% of patients seen in several uveitis series,32 and it has been reported in identical twins.46
Neovascularization of the anterior chamber angle and of the iris is seen. Although its cause is unknown, abnormalities in the iris circulation in fluorescein angiography suggest a poor arterial circulation and would support hypoxia as a possible underlying cause for the neovascularization and iris stromal changes.47,48
Posterior synechiae are extremely rare but have been reported when iris cellular nodules are present.41 Rubeosis has been noted,49 and neovascular changes have also been associated with the recognized appearance of a filiform anterior chamber hemorrhage following paracentesis of the anterior chamber. In one study this occurred in 22 of 23 patients, starting usually in the angle 180° from the site of paracentesis and producing a small hyphema in the affected eye.50
Glaucoma occurs in approximately 15% to 59% of patients with heterochromic iridocyclitis and is believed to be due to trabecular sclerosis, owing to plasma cell obstruction of the angle, or to neovascularization.33,34,51 The glaucoma is sometimes very difficult to control; if due to plasma cell infiltration, treatment with corticosteroids may be helpful. Usual medical management with timolol, miotics, and carbonic anhydrase inhibitors may be effective, but varied responses are encountered and control of pressure by these means may be temporary. An abnormal membrane in the angle has been reported visible in advanced glaucoma poorly responsive to medical management. This glaucoma seems poorly responsive to laser trabeculoplasty and usually requires some type of filtration procedure.52 Trabeculectomy and trephining have been reported as successful in controlling the pressure.52,53
Cataract surgery has been reported to have a good prognosis in a retrospective study of 29 patients reported by Smith and O’Connor54 and in a series by Franceschetti.55 Liesegang52 reported on a series of 17 patients undergoing cataract surgery. Multiple complications were experienced, and only 8 of the 17 obtained 20/40 (6/12) vision or better. Extracapsular cataract extraction and pseudophakic implantation within the capsule have been reported with good results; when successful, these procedures have provided much improved functional vision.35,56,57,58,59
Most patients with heterochromic iridocyclitis can be managed best with observation alone. Topical corticosteroids may decrease the anterior chamber and anterior vitreous cells and inflammatory debris, but when the topical corticosteroids are discontinued, the inflammatory products usually return to pretreatment levels. If there are keratic precipitates and opacities in the visual axis, vision may be temporarily improved by the use of corticosteroids. Usually, cataract is the cause of the visual loss, and prolonged use of corticosteroids may cause more problems with cataract formation and aggravate glaucoma in corticosteroid-sensitive patients. Mydriasis may be necessary if iris nodules are present and if formation of synechiae appears likely, although this is rare in this condition.
The etiology of heterochromic iridocyclitis remains obscure. Its relation to other types of heterochromia seems unclear. Damage to the sympathetic nervous system and heterochromic iridocyclitis together was substantiated in only 12 of the 1,771 cases reviewed.33 Reports of histologic changes with lymphocytes, plasma cells, and Russell bodies infiltrating the iris support an etiology of active local immunoglobulin production. It has been suggested that an immunologic etiology may be present in cases involving a local defect in B-cell regulation and formation of local immune complexes producing vascular disease, seen as chronic vascular occlusive changes and subsequent infarction, hypoxia, neovascularization, and vascular leakage. This hypothesis remains to be confirmed, but it is consistent with many observations seen in the condition, both clinically and histopathologically.35,60,61,62,63,64,65
PROGRESSIVE FACIAL HEMIATROPHY
Progressive facial hemiatrophy may sometimes be associated with a low-grade chronic iridocyclitis on the affected side. This disease has many of the characteristics of Fuchs heterochromic iridocyclitis, including the unilateral asymptomatic onset, atrophic iris with subsequent color change, keratic precipitates, cataract changes, and slowly progressive course. The cause of the hemifacial atrophy is unknown. It is sometimes accompanied by the coup de sabre lesion, and the ocular changes are believed to be due to the same processes causing the facial hemiatrophy.34
SARCOIDOSIS
Sarcoidosis is a systemic granulomatous process of unknown etiology. The incidence of clinically diagnosed sarcoid is reportedly 30 to 64 in 100,000; autopsy studies show the true incidence to be 10 times this rate, suggesting that most cases go undiagnosed.66,67 Fatigue is frequently the only symptom of low-grade systemic involvement. Diagnosis is simplified when chest films show typical mediastinal lymph node enlargement, tissue specimens of lesions show noncaseating epithelioid cell granulomas on biopsy, or systemic symptoms and disease are sufficiently characteristic to clarify the disease process. One organ system or region may be significantly involved, as demonstrated by high CD4+TH1 lymphocyte concentrations in noncaseating granulomas, but no change may be seen in the systemic T-cell populations or other systemic indications of inflammatory activity. Sarcoidosis may represent a final common pathway with common histologic pictures due to a class of poorly cleared antigens from multiple sources that trigger more favorably the CD4+TH1 lymphocyte response, then inadequate control of this response by the individuals’ immune regulation, which may explain the difficulty in defining a single etiology for this condition. Patients exposed to a variety of environmental agents, such as beryllium, pine dust, and peanut dust, present with distinctly different diseases but share a common histologic appearance to sarcoidosis. Genetic factors also no doubt play an important part in this process. Major histocompatibility complex (MHC) class I (HLA-A1, HLA-B7, HLA-B13, HLA-B15, HLA-B27, HLA-B25, and HLA-CW7) and class II (HLA-DR3) antigens are found to be increased in patients with sarcoidosis and may have protective as well as predisposing effects. The role of hyperglobulinemia and immune complexes sometimes noted also remains to be clarified.68 The unique aspect of anterior chamber-splenic handling of intraocular antigens and their disease-process considerations for sarcoidosis raise interesting possibilities for the high incidence of ocular involvement in sarcoid, but this remains uninvestigated.69,70,71,72,73
Ocular involvement is seen in approximately 27% of patients with systemically diagnosed sarcoidosis, and anterior uveitis occurs in 66% of this ocular involvement. Eye disease may be the major clinically visible sign of activity of the disease.69 Serum lysozyme,74 angiotensin-converting enzyme levels,75 gallium scanning, and conjunctival or lacrimal gland and lip biopsies76 have been recommended to help substantiate the clinical diagnosis.
Minimal inflammatory changes, such as even a few cells in the anterior chamber, are detectable in the eye. These changes elsewhere in the body might not be recognized. Diagnosis and management of this inflammatory process requires an awareness of its ocular and systemic multiple clinical presentations and variable course.68,69,77
The intraocular inflammation may be acute, but more characteristically it is chronic and usually has few symptoms until vision is affected, or until significant structural changes have occurred. It may affect all parts of the eye.78 The chronic iridocyclitis is characterized by keratic precipitates that are well defined at the edges and tend to be more concentrated in the peripheral cornea (Fig. 8). There may be an inferior ring of these precipitates visible on the endothelium at the limbus (Figs. 9, and 10) The anterior chamber contains cells and flare, and the cells have a great tendency to form cellular nodules, either as keratic precipitates or as precipitates on the iris, or at the pupil margin as Koeppe nodules (Fig. 11). The nodules tend to become the site of both peripheral anterior synechiae and posterior synechiae. The cellular reaction is usually sensitive to corticosteroids but may recur as the corticosteroids are withdrawn.69,77 Treatment responses are further obscured by the frequent tendency for sarcoidosis to undergo spontaneous remission with time, and then recur at a later date. If oral steroids are indicated, moderate to low alternate-day dosages usually control the inflammatory process and produce less systemic side effects.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


