Chordoma and chondrosarcoma represent 2 fundamentally different and challenging central skull base pathologies. Both are largely surgical diseases with varying outcomes based on completeness of resection. Adjuvant therapy is controversial, although radiation therapy is commonly employed postoperatively, and stereotactic radiosurgery is used either in primary management or treatment of local progression.
Key points
- •
Chordomas are locally aggressive tumors derived from notochord remnants.
- •
Chondrosarcomas are generally low-grade, indolent malignancies that cause morbidity through compression of neurovascular structures at the skull base.
- •
The primary treatment of both chordoma and chondrosarcoma is aggressive primary resection followed by adjuvant radiation therapy.
- •
Heavy particle therapy, such as proton beam or carbon ion, provides higher treatment doses with less toxicity and currently is most commonly recommended for chordomas and chondrosarcomas after surgical resection.
- •
Chordomas and chondrosarcomas create multifaceted clinical challenges and should be managed by multidisciplinary skull base teams with expertise in their treatment.
Introduction
Chordoma and chondrosarcoma have been grouped together historically because of the midline presentation, similar radiography, and confusion in initial pathology. However, these lesions are distinct clinicopathological entities and vary significantly in their clinical outcome. Both lesions are uncommon and represent less than 1% of intracranial lesions. Each poses a daunting challenge to effective treatment due to the perilous proximity of critical cranial nerves, the brainstem, and the skull base vasculature. To that end, both tumors have a tendency to recur locally, even after aggressive resection, especially if known residual tumor is left behind. Chordomas may metastasize with time; however, they rarely present as such. Despite these concerns, the combination of maximal resection with minimal morbidity, followed by aggressive high-dose postoperative radiation therapy, can cure most low-grade chondrosarcomas, and provides good long-term local control and quality of life in patients with chordoma and high-grade chondrosarcoma.
Chordomas are malignant primary bone tumors that are derived from notochord remnants. They were first recognized in 1856 by Lushka and concomitantly by Virchow in 1857 at autopsy. Originally thought to be of cartilaginous origin, Muller proposed these tumors were derived from notochord in 1958. The term chordoma was later suggested after these tumors were directly observed in the nucleus pulposis, incriminating their origin. They occur in the axial skeleton, where the notochord resides developmentally. Their occurrence in the axial spine can be thought of in terms of thirds, with a third arising in the skull base, spine, and sacrum 32%, 33%, and 29% of patients, respectively. It should be noted, however, that other studies support a more caudal cranial distribution of cases with less axial spine involvement. Chordoma has an incidence of 8.4 cases per 10 million population in the United States. They frequently recur locally despite aggressive surgical treatment. Although metastases do occur, these are uncommon and are not frequently existent at initial presentation. Ecchordosis physaliphora, which is also a remnant of the embryonic notochord, is a different entity and is found typically intradurally behind the clivus and anterior to the brainstem. Pathologically, ecchordosis physaliphora is virtually identical to chordoma. In the context of a patient, however, these are commonly smaller; there is complete lack of clival involvement, and they frequently follow a benign clinical course.
Chondrosarcomas are rare cartilaginous tumors that may present as low grade (World Health Organization [WHO] 1 or 2), which are far and away more common, or high grade (WHO grade 3), as well as a mesenchymal chordoma subtype. WHO grade 3 and mesenchymal subtype tumors forbear a worse prognosis. Only 1% of chondrosarcomas occur at the skull base. The cell of origin is unknown; however, typically these are thought to arise from synchodroses of the skull base, such as the spheno-occipital suture or petroclival suture. They frequently present eccentric, but do not stray far from midline. Sixty-six percent of skull base chondrosarcomas arise from the petroclival fissure; an additional 28% percent arise from the clivus proper, and a further 6% arise from the sphenoethmoidal complex. The lattermost of these is a fusion plane, which is off midline, accounting for eccentric presentation just lateral to midline. Chondrosarcomas are typically less aggressive than chordomas, rarer, and very uncommonly metastasize.
Introduction
Chordoma and chondrosarcoma have been grouped together historically because of the midline presentation, similar radiography, and confusion in initial pathology. However, these lesions are distinct clinicopathological entities and vary significantly in their clinical outcome. Both lesions are uncommon and represent less than 1% of intracranial lesions. Each poses a daunting challenge to effective treatment due to the perilous proximity of critical cranial nerves, the brainstem, and the skull base vasculature. To that end, both tumors have a tendency to recur locally, even after aggressive resection, especially if known residual tumor is left behind. Chordomas may metastasize with time; however, they rarely present as such. Despite these concerns, the combination of maximal resection with minimal morbidity, followed by aggressive high-dose postoperative radiation therapy, can cure most low-grade chondrosarcomas, and provides good long-term local control and quality of life in patients with chordoma and high-grade chondrosarcoma.
Chordomas are malignant primary bone tumors that are derived from notochord remnants. They were first recognized in 1856 by Lushka and concomitantly by Virchow in 1857 at autopsy. Originally thought to be of cartilaginous origin, Muller proposed these tumors were derived from notochord in 1958. The term chordoma was later suggested after these tumors were directly observed in the nucleus pulposis, incriminating their origin. They occur in the axial skeleton, where the notochord resides developmentally. Their occurrence in the axial spine can be thought of in terms of thirds, with a third arising in the skull base, spine, and sacrum 32%, 33%, and 29% of patients, respectively. It should be noted, however, that other studies support a more caudal cranial distribution of cases with less axial spine involvement. Chordoma has an incidence of 8.4 cases per 10 million population in the United States. They frequently recur locally despite aggressive surgical treatment. Although metastases do occur, these are uncommon and are not frequently existent at initial presentation. Ecchordosis physaliphora, which is also a remnant of the embryonic notochord, is a different entity and is found typically intradurally behind the clivus and anterior to the brainstem. Pathologically, ecchordosis physaliphora is virtually identical to chordoma. In the context of a patient, however, these are commonly smaller; there is complete lack of clival involvement, and they frequently follow a benign clinical course.
Chondrosarcomas are rare cartilaginous tumors that may present as low grade (World Health Organization [WHO] 1 or 2), which are far and away more common, or high grade (WHO grade 3), as well as a mesenchymal chordoma subtype. WHO grade 3 and mesenchymal subtype tumors forbear a worse prognosis. Only 1% of chondrosarcomas occur at the skull base. The cell of origin is unknown; however, typically these are thought to arise from synchodroses of the skull base, such as the spheno-occipital suture or petroclival suture. They frequently present eccentric, but do not stray far from midline. Sixty-six percent of skull base chondrosarcomas arise from the petroclival fissure; an additional 28% percent arise from the clivus proper, and a further 6% arise from the sphenoethmoidal complex. The lattermost of these is a fusion plane, which is off midline, accounting for eccentric presentation just lateral to midline. Chondrosarcomas are typically less aggressive than chordomas, rarer, and very uncommonly metastasize.
Presenting characteristics
Chordoma
These are the most common extradural clival tumors. The median age of presentation for skull base chordomas appears to be 60 years of age; however, there is a large range of presenting ages, including pediatric cases. Chordomas produce morbidity and mortality through local growth. Often upper clival chordomas will present with sixth nerve palsies secondary to the tumor accessing the basilar venous plexus between the periosteal and parietal dural layers producing a local compartment syndrome of dorello’s canal. However, other cranial neuropathies can be seen with cavernous sinus involvement, including third and less commonly fourth nerve palsies. Headache is another frequent presenting symptom, and in larger tumors, nasal obstruction can be seen as well as eustachian tube dysfunction. If there is brainstem compression, there may be imbalance or hemiparesis as a presenting symptom. Lower clival tumors can present with dysphagia due to mass effect or involvement of the lower cranial nerves. There is no association known between chordomas and environmental exposures or radiation induction of chordomas. There are no associated syndromes of which chordomas are a part; however, the investigation of (T) bracyury, brachyury duplication, and tuberous sclerosis complex are being investigated with respect to genetic predisposition.
Chondrosarcoma
Chondrosarcoma is less common than chordoma, particularly in the setting of skull base tumors. In combined series, the occurrence is typically one-half to one-third the frequency of chordomas. Interpretation of incidence is a bit confounding due to the fact that older combined series may have mistaken chondrosarcoma for chondroid chordomas, while now this distinction is more clear. The presenting characteristics for chondrosarcoma are similar to those stated previously for chordomas. Eccentric location, however, gives a slightly higher rate of cavernous sinus involvement, and associated diplopias are more frequent. Although chondrosarcomas typically occur in isolation, unlike chordomas, they can be associated with Paget disease, Ollier disease, and Maffuci syndrome.
Imaging
Chordomas
Imaging characteristics are fairly uniform for chordomas ( Fig. 1 ). Classically they appear hyperintense on T2 weighted MRI. On T1 weighted MRI, they are somewhat heterogeneous, with a predominantly hypointense signal. Gadolinium enhancement is variable. The authors’ own historical institutional data of over 30 skull base chordomas demonstrate minimal enhancement in 45% of cases, normal enhancement in 30% of cases, and avid enhancement in 25% of cases. Diffusion restriction is seen; however, the bony interfaces of the skull base make this finding unreliable. Thirty percent of cases demonstrate calcifications within the tumor. Dural transgression occurs 50% of the time at presentation. In terms of anatomic localization, 80% of cases demonstrate posterior clinoid involvement; 30% have occipital condylar involvement. Seventy percent of cases have cavernous sinus involvement; involvement of the upper half of the clivus occurs in 95% of cases, and the lower half of the clivus is involved in 30% of cases.
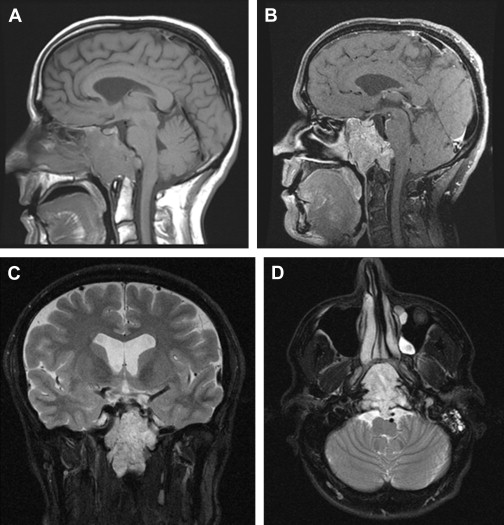
Chondrosarcomas
Imaging characteristics are similar to chordomas in that bright signal on T2 is a hallmark characteristic ( Fig. 2 ). As stated previously, chondrosarcomas are more eccentric to midline but are by no means lateral skull base lesions. Chondrosarcomas have variable World Health Organization (WHO) grades from 1 to 3; however, there does not appear to be any imaging hallmarks to differentiate low-grade from high-grade chondrosarcomas.
Pathology
Grossly, chordomas are lobulated and greyish-to-dark tumors. They can be firm with calcifications; however, in one author’s (Van Gompel) they experience, typically are gelatinous and suckable. Chordoma can come in one of 3 histologic subtypes, namely classical (conventional), chondroid, and dedifferentiated. They stain positive for brachyury, which links it to its cell of origin, the notochord. These tumors stain positive for S-100 and epithelial membrane antigen (EMA). Virchow himself first described this tumor with intracellular bubble-like inclusions, also known as physaliferous cells, which are essentially pathognomonic for this tumor. Unlike chondroid chordoma, chondrosarcomas are nonreactive for keratin and epithelial membrane antigen. Chondrosarcomas may have a gross appearance much like chordomas, but more frequently are firm, and calcifications occur within them. Histologically low-grade tumors again are difficult to distinguish from chordoma, while higher grade tumors are not, and a variant, the mesenchymal subtype, is considered a more aggressive chondrosarcoma. The grading is based on the degree of cellularity, atypia, and mitotic activity. In a large series of 200 patients, 51% were grade 1; 21% were grade 2, and 29% were a mixed grade 1 and 2. There were no grade 3 tumors seen.
Management
Surgery
Chordomas and chondrosarcomas
Both chordomas and chondrosarcomas should be treated by a multidisciplinary skull base team including neurosurgery, otolaryngology, medical oncology, and radiation oncology, with participation also coming from endocrinology, ophthalmology, and rehabilitation ( [CR] and [CR] ). Although en bloc resection is frequently the goal in the sacrum and the spine, rarely can a chordoma of the skull base be taken out in such a manner. Philosophy regarding the removal of these tumors has greatly evolved in recent decades, with resection in a piecemeal fashion considered to be oncologically sound. Frequently, in older series, simple biopsy was performed with intralesional curettage. The recent advent of skull base techniques and even more recent cultivation of endoscopic techniques have permitted more thorough removal of tumor with less postoperative morbidity. Certain authors have taken this to the extreme and advocate resection of as much of the clivus as possible during primary surgery with potential staged operations to achieve this, citing the frequency of recurrence as the primary reason. The current authors’ stance is similar in that all boney involvement and adjacent bone that can be safely taken should be; however, cavernous sinus involvement often precludes complete resection, and few authors have advocated carotid resection because of high morbidity. In turn, adjuvant radiation can be used to treat residual tumor. It should be noted that aggressive surgical series document a higher rate of carotid injury, which may indicate that chordomas themselves locally invade the carotid artery, potentially leading to higher carotid artery injuries in the operating room. This also may be related to the aggressive attempts at resection. The authors advocate a plan to deal with intraoperative carotid bleeding, and in select cases perform preoperative balloon occlusion studies in preparation for such a catastrophic vascular injury.
Lesions of the lower clivus pose a similar dilemma in that complete boney resection in this area may impart occipital cervical instability. In younger and healthier patients, however, complete resection with concomitant posterior fusion may be an option to obtain better margin status. Typically, if the dura is not invaded, it is left in situ as a barrier; however, if there is dural transgression, the authors advocate dural resection with a negative margin goal. Brainstem involvement is managed with resection of the capsule of the tumor, but brainstem margins are not taken. In cases of cranial nerve involvement, hypoglossal being the most frequent conundrum, resection of this nerve is only undertaken if there is an opportunity for complete resection. The authors do not advocate bilateral hypoglossal sacrifice if there is bilateral involvement due to the morbidity of such a resection to the patient. The sixth nerve is also frequently involved in Dorello canal; however, in such circumstances, the authors typically perform decompression, resecting the tumor around the nerve but not the nerve itself. Although the approach to each tumor is variable depending on size and location, an anterior approach is favored, because most of these tumors arise midline in the clivus. Endoscopic endonasal approaches generally allow access from the frontal recess to the base of C2. Advanced endoscopic approaches, such as the transpterygoid procedure, the extradural pituitary transposition ( Fig. 3 ), the petrous apex approach ( Fig. 4 ), and the far medial approach ( Fig. 5 ) can be employed to achieve complete resection of tumor.



