Cholinergic Pharmacology
Joel S. Mindel
BASIC PHARMACOLOGY
The classic cholinergic systems are located at neuron-neuron synapses, at neuron-striated muscle fiber junctions, and at neuron-smooth muscle fiber junctions.
Impulses are transmitted across the spaces separating these structures by acetylcholine. A 45-kd glycoprotein, called the cholinergic neuronal differentiation factor, seems to play a role in determining which embryonic neurons will develop acetylcholine synthesizing properties.1,2
The earliest pharmacologic studies were of those tissues innervated by the parasympathetic nervous system. Dixon3 pointed out that the application of muscarine produced effects that were identical to those produced by the stimulation of the vagal nerve. Loewi4 demonstrated that vagal nerve stimulation released a substance that he subsequently identified as acetylcholine. Dale5 was the first to suggest that the action of acetylcholine was terminated by an enzyme, an esterase, that hydrolyzed the molecule.
Cholinergic systems consist of a number of subcellular components: choline acetyltransferase; acetylcholine; sodium-dependent, high-affinity choline uptake system; clear vesicles; acetylcholinesterase; and nicotinic and muscarinic receptors.
CHOLINE ACETYLTRANSFERASE
The gene for choline acetyltransferase is located on the long arm of human chromosome 10.6 Within its length it also contains the gene that encodes the protein responsible for transporting acetylcholine into the vesicle. Although these two genes overlap, they do not share similar protein sequences.7
Choline acetyltransferase synthesizes acetylcholine from choline and acetyl-coenzyme A. Choline acetyltransferase is found in the soluble cytoplasm and attached to the outer membrane of acetylcholine-containing vesicles. It is not found within these vesicles.8,9 The soluble form predominates at more physiologic pH.10 The binding of choline acetyltransferase to vesicle membranes is reversible and presumably due to ionic attraction between positively charged choline acetyltransferase and negatively charged membrane. For maximum enzymatic synthesis of acetylcholine, higher concentrations of choline than of acetyl-coenzyme A are required. For example, in human brain, the Michaelis’ constant (Km) for choline is 510 μmol/L and the Km for acetyl-coenzyme A is 11 μmol/L.11 Imidazole-containing compounds, such as pilocarpine, stimulate in vitro enzymatic synthesis of acetylcholine.12,13 The possibility of small amounts of nonenzymatic production of acetylcholine by imidazoles has also been raised.14
ACETYLCHOLINE
Acetylcholine is found in both the cytoplasm and vesicles of cholinergic nerve endings. When high concentrations of cytoplasmic acetylcholine exist, choline kinase is stimulated. Choline kinase phosphorylates choline and diverts it away from acetylcholine synthesis and toward phospholipid synthesis.17 In this way, acetylcholine may regulate the availability of its own substrate. The acetylcholine-containing vesicles are 30 nm to 60 nm in diameter and are electron transparent (i.e., “agranular”). They contain high concentrations of acetylcholine (approximately 47,000 molecules per vesicle [520 mmol/L]) and adenosine triphosphate (ATP) (approximately 17,000 molecules [170 mmol/L]); guanosine triphosphate (GTP) is present in 20 mmol/L concentration.18
When the neuron is at rest, acetylcholine is released spontaneously from the cytoplasm by a non-calcium-dependent mechanism. The amounts of cytoplasmic acetylcholine leaked are inversely regulated by neuronal membrane ATPase activity (i.e., the greater the ATPase activity, the lower the amounts). Evoked release of acetylcholine occurs from vesicles and depends on the presence of extracellular calcium.19,20,21
Recycling of a vesicle (i.e., the time from acetylcholine release to refilling and re-release) appears to take approximately 1 minute.22 This cycle can be divided into eight steps23:
Docking (vesicle contacts internal surface of synaptic axon membrane)
Priming (conformational change allowing calcium to trigger exocytosis of acetylcholine)
Exocytosis (calcium produces release of acetylcholine into the synaptic space)
Endocytosis (vesicle closes)
Translocation (vesicle is moved internally)
Endosome fusion
Neurotransmitter uptake
Translocation (vesicle returns near synaptic axon membrane)
A number of proteins in the membrane of the synaptic vesicle have been identified (e.g., synaptophysin and synaptotagmin).24,25 The acetylcholine transporting protein identified on chromosome 10 contains approximately 40% of the amino acid sequences contained by these two vesicular proteins and is believed to have 12 transmembrane spanning domains. Thirty or more low-molecular-weight GTP-binding proteins, called Rab proteins, are involved in directing the vesicle through the cycle. One cluster, called the synaptosecretosome, triggers acetylcholine exocytosis; the key protein in this group is called mediatophore. Other proteins link the vesicle to the synaptic membrane. Some of these have channel structures that could form the route taken by released acetylcholine on its way to the synaptic space. This raises the possibility that direct or full fusion of the vesicle with the synaptic axon membrane is not needed.26,27 A specific inhibitor of acetylcholine uptake into cholinergic vesicles, vesamicol, has been identified. Botulinum toxins attack some of the proteins linking the synaptic vesicle to the axon membrane and prevent calcium from triggering functional acetylcholine release.28,29,30
CHOLINE UPTAKE
Acetyl-coenzyme A is synthesized in neuronal mitochondria from pyruvate, by pyruvate dehydrogenase, and transferred into the cytoplasm for acetylcholine synthesis. Choline is either synthesized by the neuron or obtained from the circulation.31 Neurons can synthesize choline by methylation of phosphatidylethanolamine or ethanolamine.32,33 Phospholipase D hydrolyzes phosphatidylethanolamine to phosphatidic acid and choline.34,35 Choline can be taken up by a neuron over its entire surface if a high concentration (i.e., Km = 10 to 100 μmol/L) is present.36 This system is referred to as the low-affinity choline uptake system and probably plays little, if any, physiologic role. However, a sodium-dependent, high-affinity choline uptake system (i.e., Km = 1 to 10 μmol/L) is located on the synaptic nerve endings.37 The high-affinity system becomes more active when the neuron is releasing acetylcholine and/or when it is synthesizing more acetylcholine.38,39,40 Hemicholinium blocks the high-affinity choline uptake system. Although the choline transported into the nerve terminals is rapidly acetylated, there does not appear to be a direct linkage between the high-affinity system and choline acetyltransferase.41
RECEPTORS
There are two types of cholinergic receptors: muscarinic and nicotinic. Classically, these receptors were thought to exist only on the distal sides of the neuron-muscle fiber and neuron-neuron junctions (i.e., postjunctional receptors). A large amount of evidence indicates that they exist on the proximal sides of these junctions as well (i.e., the neuron terminal releasing acetylcholine has prejunctional receptors). Postjunctional receptors are effector receptors. Prejunctional receptors are involved in feedback stimulation and inhibition of transmitter release.
Postjunctional Receptors
NICOTINIC. Nicotinic receptors respond to acetylcholine and nicotine but not to muscarine. They are found in the central nervous system, in ganglia, and on striated muscle fibers. The specialized receptor area of a striated muscle fiber is termed the myoneural junction. This area is marked by infolding and convolutions of the muscle fiber membrane and a significant increase in the density of acetylcholinesterase molecules.
MUSCARINIC. Muscarinic receptors respond to acetylcholine and muscarine but not to nicotine. These receptors are found in the central nervous system, in ganglia, and on smooth muscle fibers. The junction of a parasympathetic neuron with a smooth muscle fiber is not highly specialized. The two are separated by a relatively wide distance, ranging from 15 to 1900 nm. This has functional significance (i.e., the narrower the cleft, the greater the concentration of transmitter at both prejunctional and postjunctional receptors). Junctional clefts in the iris sphincter are approximately 15 nm. Muscarinic reception may occur at those sites on smooth muscle fiber membranes where electrondense structures, 5 to 6 nm thick, are found.42 Muscarinic receptors have a latency of 100 to 500 msec, in contrast to nicotinic receptors, which have a latency of 30 to 100 msec. However, it does not appear that this latency is simply a function of the distance between nerve ending and receptor.43
Nicotinic Receptor Structure. Nicotinic receptors have a molecular weight of approximately 250 kd. Each receptor consists of five glycopeptide chains (two of which are identical): two alpha, one beta, one delta, and one epsilon.44 Some species variations exist.45 Human fetal receptors are found diffusely along the muscle fiber membrane and have a gamma subunit instead of an epsilon. When stimulated, fetal receptors provide a membrane depolarization that has a longer duration and fetal receptors are less readily desensitized by prolonged agonist exposure. Conversion to the adult form occurs upon innervation when the receptors become clustered at synapses.46 Extraocular muscles are an exception in that some of their muscle fibers retain the fetal type of receptor.47,48 The fetal receptor is not found in the human adult levator palpebrae superioris of the lid.49 In rats, all of the myoneural junctions of the multiply innervated (en grappe) extraocular muscle fibers, and a limited number of the singly innervated (en plaque) extraocular muscle fibers, co-express both the adult and fetal forms of the receptor.50
Each glycopeptide subunit of the receptor spans the entire length of the receptor, and the five subunits form a pentagon around a central ion channel, which also runs the entire 11-nm length of the receptor.51 The receptor protrudes approximately 5.5 nm on the extracellular side of the cell membrane and approximately 1.5 nm on the intracellular side. The ion channel opening is widest extracellularly and then narrows as it passes through the cell membrane.52
The binding site for cholinergic drugs on striated muscle nicotinic receptors is, at least in part, on the extracellular portion of the alpha subunit in a region containing two adjacent cysteines.53,54,55 In neuronal nicotinic receptors, the binding site may be on the beta subunit.56 Because there are two alpha subunits, there are two binding sites for acetylcholine on nicotinic muscle receptors. Both binding sites must be bound to elicit a response.57 One is a high-affinity site and the other is a low-affinity site; the differences in affinity are conferred by the gamma and delta subunits where they contact the alpha subunits. The gamma subunit confers high affinity and the delta subunit confers low affinity.58
The carbonyl side of the acetylcholine molecule reacts with the nicotinic receptor, whereas the methyl side reacts with the muscarinic receptor.59 The acetylcholine molecules bind in a cooperative manner (i.e., binding of one acetylcholine molecule increases the affinity of the receptor to bind the second molecule).
Activating the receptor opens the ion channel for approximately 1 msec and produces a membrane depolarization of approximately 0.2 μV.60 The length of time the channel is open depends on the agonist (e.g., acetylcholine = 1 msec; carbachol = 0.33 msec; and succinylcholine = 0.23 msec). The nicotinic ion channel is almost equally permeable to Na+ and K+ ions but impermeable to Cl- ions. Larger monovalent cations pass through the channel more readily than do smaller ones.61 This is reversed for divalent cations (i.e., smaller ones pass through more readily). Differences in potencies between agonists are due to affinity for the receptor and efficacy for opening the channel. Carbachol has a channel-opening efficacy that is about two thirds that of acetylcholine. Nicotinic receptors outside the synaptic area stay open about three times as long as those in the synaptic area. The activated ion channels of singly innervated (i.e., en plaque or fast) muscle fibers have characteristics similar to those of multiply innervated (i.e., en grappe or slow) muscle fibers.62
Competitive nicotinic antagonists interfere with acetylcholine’s binding onto the alpha subunits. Noncompetitive antagonists may allow the ion channel to open, but then block it.60 The action of a specific drug may be complex. For example, curare is classified as a nondepolarizing, noncompetitive nicotinic antagonist. It binds to the receptor at a site other than where acetylcholine binds (i.e., it is not competitive with acetylcholine at this point). Once bound, curare exerts its main action by competing with acetylcholine for the site on which the latter acts to open the ion channel.63 Thus, there are competitive and noncompetitive aspects to curare’s action. Furthermore, high concentrations of a competitive agonist, such as concentrations of acetylcholine above 300 μmol or carbachol above 1 mmol, can enter and transiently occlude the ion channel.64 Decamethonium and succinyldicholine act primarily by blocking open channels. Such channel blockers cannot be antagonized by cholinesterase inhibitors because the latter result in prolonged and increased endogenous acetylcholine concentrations, which, in turn, stimulate the receptor to keep the ion channel open and the antagonist blocking it.60
Muscarinic Receptor Structure. Whereas a nicotinic receptor consists of multiple subunits arranged to form an ion channel, a muscarinic receptor is a single glycoprotein that activates a G protein (i.e., a guanine nucleotide binding protein). Muscarinic receptors belong to a G protein-linked “superfamily” of receptors that includes α1-, α2-, and β-adrenergic receptors and serotonin receptors. All members of the family have an undulating ringlike structure with seven hydrophobic transmembrane spanning groups. These transmembrane groups are linked by alternating cytoplasmic and extracellular loops.65,66 The possibility exists that some muscarinic receptors consist of two subunits.67,68 Different muscarinic receptors have approximately 40% identical amino acid sequences. Within the superfamily, this falls to approximately 4%, with only 19 amino acids conserved among all known members.
Five different muscarinic receptor genes and their different receptor products have been identified. These are commonly designated as m1, m2, m3, m4, and m5. Pharmacologic evidence for the functional existence of these receptors in tissues exists for only four of them, designated M1, M2, M3, and M4.69
In man, the m1 receptor consists of 460 amino acids; m2 has 466 amino acids, m3 has 590 amino acids, and m4 has 471 amino acids.70,71 The lack of highly specific drugs for each receptor subtype has hampered the identification of the corresponding functional tissue receptors. M1 receptors have a high affinity for the antagonists pirenzepine and 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP) and a low affinity for the antagonists methoctramine and himbacine. M2 receptors have the reverse affinities. M3 receptors have a high affinity for 4-DAMP but a low affinity for pirenzepine.72 Carbachol has a much greater affinity for M2 than for M1 or M4 receptors.65 The formation of an ionic bond between the amine group of the agonist or antagonist and the aspartic acid on the extracellular loop of the third transmembrane spanning group seems to be a feature of all muscarinic receptors.73 However, there may be more than just one drug binding site.74 The third cytoplasmic loop is the major determinant for G protein coupling selectivity.75,76
Prejunctional Receptors
Since 1959, hypotheses have existed to explain why nicotine produces adrenergic effects when applied to sympathetic nerve endings.77 Whereas nicotine causes an increase in norepinephrine release, muscarine and pilocarpine inhibit norepinephrine release. Pilocarpine’s effect can be blocked by atropine. There is now a large body of evidence showing that receptors are also present on nerve endings. For example, there is markedly less binding of muscarinic drugs after sympathetic nerve endings have been destroyed by 6-OH dopamine.78 These presynaptic receptors are believed to play a role in feedback control of receptor release. In this manner, release of acetylcholine by parasympathetic nerve endings could alter adrenergic activity. It appears that the muscarinic receptor is activated at lower acetylcholine concentrations than is the nicotinic, but when both are stimulated maximally, the effect of the nicotinic receptor predominates. Thus, when sympathetic nerve endings are bathed in low concentrations of acetylcholine, there is little or no norepinephrine release. The addition of atropine causes increased norepinephrine release. At higher acetylcholine concentrations, additional norepinephrine is released as the nicotinic receptors are stimulated. The muscarinic receptors are believed to be of greater physiologic importance. The existence of prejunctional receptors may explain why systemic administration of muscarinic drugs in vivo causes blood vessel dilatation even though blood vessels are innervated almost exclusively by adrenergic fibers.
Muscarinic presynaptic receptors have been found on acetylcholine-releasing neurons of striated muscle.79 These may mediate a decrease in the release of neuronal acetylcholine by a feedback mechanism.80
A variety of prejunctional receptors, besides nicotinic and muscarinic ones, exist on acetylcholine-releasing neurons. Parasympathetic neurons have prejunctional alpha receptors that, when stimulated, may inhibit acetylcholine release.81 Another prejunctional receptor appears to be sensitive to the purine nucleotide coenzyme A. Coenzyme A is a major product of acetylcholine synthesis. Coenzyme A has been reported to act as a presynaptic inhibitor of acetylcholine release and could modulate cholinergic activity.82 Prejunctional prostaglandin receptors may also exist on parasympathetic nerve endings. Prostaglandins are released during parasympathetic activity83 and are reported to both facilitate and inhibit acetylcholine release.84,85 However, there are also reports that prostaglandins have no effect on parasympathetic motor neurons.86,87
INTRACELLULAR EFFECTS OF MUSCARINIC AGONISTS. Activated muscarinic receptors couple to specific intracellular proteins. This GTP-requiring coupling results in enzyme activation. Depending on the receptor subtype, phospholipase C may be activated and/or adenylcyclase activity increased or inhibited. Different effects have been reported by different laboratories.71,73 A consensus evaluation is that m1, m3, and m5 receptors strongly activate phospholipase C; m2 and m4 receptors strongly attenuate adenylcyclase activity while, to varying degrees, weakly increasing phospholipase C activity. Phospholipase C hydrolyzes phosphatidylinositol 4,5-biphosphate into myoinositol 1,4,5 triphosphate- and 1,2-diacylglycerol.89 In muscle cells, the most important intracellular response ultimately produced may be release of intracellular stores of calcium resulting in cell membrane depolarization and, as a secondary event, entry of extracellular calcium.90
Supersensitivity
In normally innervated striated muscle, nicotinic receptors are found almost exclusively at the motor endplate region (i.e., the density of receptors at myoneural junctions is more than 2500 times that at extrajunctional sites).91 Slow-twitch muscle fibers, such as the multiply innervated en grappe extraocular muscle fibers, tend to have more extrajunctional receptors than singly innervated fast-twitch fibers.92,93 Extrajunctional receptors have a short half-life of approximately 19 hours, whereas those in the myoneural junction tend to be more stable with a half-life of approximately 12 days.94
Denervation reduces the normal motor endplate nicotinic receptor half-life to approximately 3 days.95 These receptors are subsequently replaced by receptors having a shorter half-life, similar to that of the extrajunctional receptors of innervated muscle.96
When skeletal muscle is denervated, the number of receptors increases and they spread out from the myoneural junction.97,98 Approximately 3 to 4 days after denervation, receptors appear outside the original synaptic area. Their number is maximum in approximately 2 weeks. After 6 to 8 weeks of denervation, extrajunctional receptors are no longer found in the degenerating fibers.99 Electrical stimulation prevents this spread of receptors and reduces supersensitivity to acetylcholine.100,101,102 Disuse can produce a small increase in the number of receptors even if innervation is present.103,104
Denervation sensitivity in nicotinic structures may be associated not only with an increase in receptors but also with a decrease, or increase, in acetylcholinesterase activity.105,106,107 A decrease would explain why sensitivity to acetylcholine increases by a factor of 1000 while the number of receptors increases only 20- to 30-fold.108
The mechanism of parasympathetic muscle fiber supersensitivity is not as clear. There is some evidence that the number of receptors increases, although not dramatically.109 However, binding studies are unable to distinguish between presynaptic and postsynaptic receptors. Thus, the number of postsynaptic receptors could increase and the number of presynaptic receptors could decrease with little overall change in total binding. There is evidence that a fall in acetylcholinesterase activity occurs.110 Additional causes of supersensitivity for which there is evidence are (1) the receptors formed after denervation may be qualitatively different111 and (2) denervation may lead to a less stable muscle membrane, as shown by an increased sensitivity to extracellular ions such as potassium.112
Subsensitivity
Sustained activation of nicotinic or muscarinic receptors can result in subsensitivity (i.e., decreased muscle contraction on re-exposure to the agonist).113 In nicotinic receptors, desensitization has been associated with increased protein kinase A activity.114
In smooth muscle, prior atropine treatment prevents this effect. Pilocarpine stimulation does not reduce the number of receptors.115 Short-term (less than 1 hour) desensitization seems due to decreased agonist binding and/or inactivation of the intracellular coupling to guanylate cyclase. Longer-term desensitization seems due to decreased number and affinity of receptors. The half-life for receptor disappearance is 4 hours. The return of receptor sites (half-time = 6 hours) when acetylcholine is removed is more rapid than the return of their function (half-time = 16 hours).116
Muscarinic receptor desensitization is associated with two phenomena. One is phosphorylation of the receptor by a G protein-coupled kinase, which allows binding by a member of the arrestin family of proteins. Arrestin binding prevents coupling of the receptor with other G proteins. The major site for phosphorylation is on the receptor’s third intracellular loop; there appear to be multiple phosphorylation sites on this loop.117 The other desensitization phenomenon is sequestration (i.e., internalization of the receptor away from the cell surface). Although sequestration may be facilitated by phosphorylation,118 the two phenomena occur by independent mechanisms.119
ACETYLCHOLINESTERASE
Acetylcholinesterase has been found in all cell membranes examined, even red blood cells.120 Its function in cholinergic systems is to terminate the action of acetylcholine by hydrolyzing the transmitter to choline and acetic acid. Acetylcholinesterase can also hydrolyze the peptide bonds found in neuropeptides, such as substance P and enkephalins.121 In addition, the acetylcholinesterase-containing protein molecule may also contain other enzymatic functions, such as Na-K activated ATPase.122 It may, therefore, play a role in the membrane permeability changes that occur after acetylcholine-receptor interaction.123
Acetylcholinesterase first binds the positively charged acetylcholine molecule at a negatively charged site, the “anionic” site. This anionic site is located in a cleft in the enzyme. The ionized rings of aromatic amino acids line the cleft and form the anionic site. Close to the anionic site is the catalytic “esteradic” site where hydrolysis occurs.124 This esteradic site is also located in the cleft, approximately 2 nm deep (i.e., halfway through the full thickness of the enzyme). The esteradic site itself consists of a triad of amino acids: a serine, a histidine, and a glutamic acid.125 After the anionic site has attracted, bound, and oriented acetylcholine, the esteradic site hydrolyzes acetylcholine in the sequence: acetylcholine-acetylcholinesterase → acetyl-acetylcholinesterase + choline → acetylcholinesterase + acetic acid + choline.
In addition, there is another anionic site on acetylcholinesterase located more distal from the esteradic site.126 It may help guide acetylcholine to the active cleft in the esterase. Binding to this secondary anionic site by some drugs seems to inhibit the action of the esteradic site, which may account for why large concentrations of acetylcholine inhibit acetylcholinesterase. However, other drugs (e.g., atropine), upon binding to the secondary anionic site, appear to activate the esteradic site. Edrophonium (Tensilon), procaine, curare, tetracaine, and decamethonium only seem to bind the anionic site that is near the esteradic site.127
Edrophonium, like acetylcholine, has a quaternary amine that interacts with an indole ring of a tryptophan molecule located at the primary esteradic site.128,129 This places the meta hydroxyl group of edrophonium between the serine and histidine in the catalytic site, further blocking acetylcholine’s approach.130 The phosphorylating and carbamylating cholinesterase inhibitors form covalent bonds with the serine in the esteradic site.131 Demecarium has additional potency because it inhibits the esteradic site and because its two nitrogen atoms interact with both the primary and secondary anionic sites, which are 1.4 nm apart.132
Acetylcholinesterase can be inhibited by a number of drugs. The result of this inhibition is that endplate potentials become more prolonged because individual acetylcholine molecules can repeat their effect.133 Inactivation of acetylcholinesterase causes the half-decay times of the endplate currents to increase from 1.5 to 2 msec to 3 to 10 msec. Acetylcholinesterase inhibitors may also act by competing with acetylcholine for nonspecific binding sites, thereby increasing unbound levels of acetylcholine.134
Acetylcholinesterase inhibition by organophosphorus compounds is irreversible unless the esteradic site is rapidly dephosphorylated by nucleophilic agents such as pyridine oximes.135 If reactivation is not rapid, the phosphorus group can no longer be removed by nucleophilic agents. The amount of acetylcholinesterase that can be reactivated decreases exponentially with time because of a process called “aging.”136 Aging occurs because of the release of an oxygen-bounded alkyl group from the enzyme-phosphorus complex.137,138 The rate of aging depends on the organophosphorus compound used.139 It can be retarded by prior administration of quaternary amines140,141 and imidazole-containing compounds, of which pilocarpine is an example.142,143 By using organophosphorus compounds, an estimate can be made of the rate of acetylcholinesterase synthesis (e.g., 50% of brain acetylcholinesterase activity has recovered 1.2 to 5.3 days after subcutaneous injection of soman).144 Cholinesterase inhibitors differ in their abilities to penetrate the cell membrane. Those that can penetrate will inhibit cytoplasmic as well as cell membrane acetylcholinesterase. Diisopropyl fluorophosphate (DFP, isoflurophate) is uncharged and can penetrate the cell membrane well. The recovery of cholinesterase activity is therefore longer (7 to 11 days) after DFP administration.145,146
Cholinesterase exists in two forms, asymmetric and globular.147 The asymmetric form consists of 1 to 3 catalytic subunits attached to a collagen-like tail 500 nm in length. Each of these catalytic subunits is a tetramer made up of 4 sub-subunits. Thus, asymmetric cholinesterase may contain 4, 8, or 12 sub-subunits. Each sub-subunit has a mass of 80 to 90 kd. Sub-subunits are joined together in pairs by a single covalent disulfide bond.148,149,150 Two of these pairs are held together by hydrophobic forces to create a tetramer. The collagen-like fibrous tail is a triple helix that anchors the enzyme to the cell membrane. At synaptic sites, the acetylcholinesterase is the 3-tetramer asymmetric form. Denervation of striated muscle results in a decrease of this form.151
The globular form consists of 1, 2, or 4 of the sub-subunits. Globular forms are attached to the cell membrane in various ways, one of which is by attachment to phosphatidylinositol. Human butyrylcholinesterase (also referred to as pseudo- or serum cholinesterase) is 95% globular tetramers; the remaining 5% monomers and dimers are believed to be degradation products.
The substrate specificities of acetyl- and butyrylcholinesterases are not due to their asymmetric or globular form. For example, small amounts of globular tetramers of acetylcholinesterase can be detected in serum. Similarly, although asymmetric cholinesterases are found only in peripheral nerve and muscle, both asymmetric acetylcholinesterase and asymmetric butyrylcholinesterase can be detected. Hybrids of acetylcholinesterase and butyrylcholinesterase exist. The tails of pure asymmetric acetylcholinesterase and pure asymmetric butyrylcholinesterase are significantly different in structure.152
Smooth-muscle membranes contain the 3-tetramer asymmetric form of acetylcholinesterase as well as other forms. Although the absence of discrete myoneural junctions results in there being relatively less of the 3-tetramer asymmetric form compared with striated muscle fibers, it is interesting that the smooth muscles with the highest percent are the mammalian iris and ciliary body.153
OCULAR CHOLINERGIC SYSTEMS
In the past, a cholinergic system was defined as consisting of a triad: choline acetyltransferase, acetylcholine, and muscarinic or nicotinic receptors. If these three components were found in proximity, one of three types of cholinergic systems could be identified: (1) a parasympathetic motor nerve ending-smooth muscle junction, (2) a nicotinic nerve-striated muscle “myoneural” junction, or (3) a neuron-neuron synapse. However, the definition of a cholinergic system is no longer clear-cut. Are prejunctional nicotinic and muscarinic receptors on adrenergic nerve endings part of a cholinergic system? Is a structure without innervation (such as the placenta) that contains large amounts of choline acetyltransferase and acetylcholine, cholinergic?154 The following discussion will present evidence for the existence of cholinergic components in ocular tissues. It should also be remembered that pharmacologic doses of drugs may act at sites not affected by physiologic doses (i.e., proteins that do not normally act as receptors may respond to high concentrations of drugs).
CHOLINE ACETYLTRANSFERASE
The corneal epithelium,155 iris, ciliary body,156 and retina156 of some mammals, including humans, contain large amounts of choline acetyltransferase activity.157 Nonmammals, such as turtles, have retinal photoreceptors containing choline acetyltransferase activity.158 Ross and McDougal159 have found that the inner plexiform layer contains the highest level of enzyme activity in mammalian retina.
ACETYLCHOLINE
Large quantities of acetylcholine have been detected in the corneal epithelium but not in the stroma or endothelium.160,161,162 Investigations have attempted to link corneal acetylcholine with ion transport163 and touch sensation.164 Acetylcholine activity has also been detected in iris, ciliary body, retina, and choroid.165,166,167 Dowling and Boycott,168 using electron microscopy, have observed vesicles in retina that are similar to those associated with acetylcholine storage.
ACETYLCHOLINESTERASE
Significant levels of acetylcholinesterase have been found in the corneal epithelium, with lesser amounts in stroma.169,170,171 Fresh primary aqueous and vitreous humors contain virtually no cholinesterase activity, but secondary aqueous or aqueous humor removed after eyes have been refrigerated 5 to 6 hours does contain increased activity, mainly that of butyrylcholinesterase.172 The increase in secondary aqueous activity is attributed to breakdown of the blood-aqueous humor barrier and to penetration of serum cholinesterase. The increase in cholinesterase activity on storage is attributed to postmortem cellular autolysis of tissues bordering the ocular fluids. Human sclera, iris, ciliary body, retina, choroid, and optic nerve also contain acetylcholinesterase.173,174 Most of the iris enzyme activity is in the sphincter region.175 In mammalian retinas, most acetylcholinesterase activity has been found in the amacrine cells of the inner plexiform layer.176,177,178 Low concentrations of acetylcholinesterase, limited to the anterior capsular region, have been found in human lenses.179 No cholinesterase activity has been found in retinal blood vessels.180
RECEPTORS
Muscarinic receptors have been identified in rat extraorbital lacrimal gland.181 Their existence in corneal epithelium has been difficult to establish. They were not found in preparations of disrupted rabbit corneal epithelial cells182,183 but were found in cultures of rabbit,184,185,186 bovine,187 and human188 epithelium. Multiple subtypes of muscarinic receptors have been identified in rabbit corneal endothelium.186,190 Messenger RNA for m3 receptors has been found in human corneal epithelium, corneal endothelium, trabecular meshwork, anterior lens epithelium, iris epithelium, ciliary epithelium, and ciliary muscle.191 Muscarinic receptors are in mammalian iris sphincter and, to a lesser degree, iris dilator muscle.192,193,194,195 These latter receptors may be part of a reciprocal cholinergic innervation.196 Whereas mammalian iris contains muscarinic receptors, avian iris contains nicotinic receptors.197
M3 muscarinic receptors are present in rabbit,198,199 bovine,200 and human iris sphincters.201 M1 receptors are present in ciliary processes.202 M3 receptors predominate in human ciliary muscle,201 although the M2 subtype is also present.191 In both the circular and longitudinal portions of the ciliary muscle there is strong expression of m2, m3, and m5 messenger RNAs; the m2 and m3 messenger RNAs are more prominently expressed in the circular portion.203 M3 receptors have also been identified in human trabecular meshwork cells.204
In mammalian retina, there is evidence that cholinergic receptors are present.205,206,207 Muscarinic receptors are present in the human inner plexiform layer, and nicotinic binding sites have also been identified.208 In nonmammalian retina, nicotinic binding sites have been found in bipolar cell and amacrine cell synapses of the inner plexiform layer.209
CHOLINE UPTAKE
The corneal epithelium appears to have a low-affinity choline uptake system and not the high-affinity system associated with cholinergic nerve endings.210 The retina, however, does have a high-affinity choline uptake system.211,212,213 It is assumed that it is located in the membranes of the inner plexiform cells that synthesize acetylcholine. In addition, mammalian photoreceptor cells and lenses, which do not synthesize acetylcholine, also have a high-affinity choline uptake system.214,215 Thus, cells that use large amounts of choline, as would be needed for phospholipid synthesis, may contain the high-affinity system.
CHOLINERGIC NEURONS
There are difficulties with each of the techniques used to determine which ocular neurons are cholinergic.
Denervation Studies
One method is to remove the cholinergic innervation to ocular structures and examine histologically for neuronal loss. Unfortunately, this begs the question because the experimenter first defines, a priori, which nerve trunks are cholinergic. For example, the cornea has been examined for parasympathetic innervation by removal of the ciliary ganglion.216 However, a variable number of trigeminal nerve sensory fibers pass through the ciliary ganglion without synapsing.217,218 Loss of these, if detected, would be incorrectly interpreted as evidence of corneal cholinergic innervation.
Acetylcholinesterase Histochemistry
The assumption has been made that acetylcholinesterase is a marker of cholinergic neurons. However, it can be found by biochemical assay in all cells. Laties and Jacobowitz219 identified cholinergic innervation of the iris sphincter and ciliary body on the basis of cholinesterase stains. They were probably correct, but the ubiquitous distribution of the enzyme reduces the specificity of this approach.
Clear Vesicles
Nomura and Smelser220 identified clear vesicles in the nerve terminals of iris dilator muscle, iris sphincter muscle, and trabecular meshwork. They assumed that these contained acetylcholine and were cholinergic. Although acetylcholine is stored in clear vesicles, it is not as certain that all clear vesicles contain acetylcholine.
OCULAR CHOLINERGIC PHARMACOLOGY
NICOTINIC DRUGS
Direct-Acting Nicotinic Agonists
This class of drugs does not play a role in ophthalmology.
Indirect-Acting Nicotinic Agonists
Cholinesterase inhibitors increase cholinergic activity in both muscarinic and nicotinic structures. With two exceptions, these drugs are used by the ophthalmologist for their muscarinic effects. One exception is their use in the diagnosis of myasthenia gravis.
Intravenous edrophonium (Tensilon) and, to a lesser extent, intramuscular neostigmine221 have been the cholinesterase inhibitors used most frequently.
Edrophonium (Fig. 1) is a short-acting cholinesterase inhibitor. It carries a positive charge, enabling it to bind with the negatively charged “anionic” site of acetylcholinesterase. The bulky cyclic structure of edrophonium physically blocks the approach of acetylcholine to the “esteradic” site. In addition, electrostatic hydrogen bonding of the hydroxyl group of edrophonium causes inactivation of the esteradic site. Acetylcholine hydrolysis is inhibited because it must compete with edrophonium for both the “anionic” and “esteradic” sites. Edrophonium’s binding to both enzyme sites is weak and short lived.
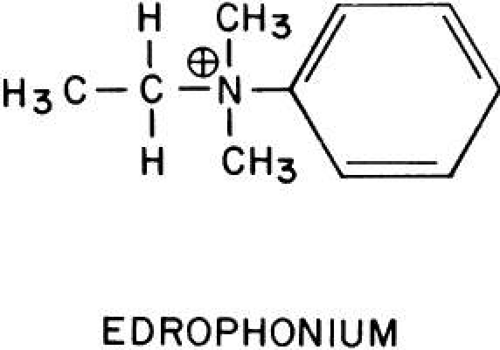 Fig. 1. Edrophonium has a quaternary amine, giving it a positive charge. It will react with the anionic site on cholinesterase. However, edrophonium will not react with the esteradic site because it has no ester bond. |
The side effects of intravenous edrophonium are the result of its action on muscarinic structures (bowel, bladder, and heart). They include abdominal cramping, urinary incontinence, bradycardia, and cardiac arrest.222,223 However, the diagnostic value of edrophonium in myasthenia gravis resides in its nicotinic action: the striated musculature contracts more vigorously. Therefore, the side effects of edrophonium can be prevented without interfering with the drug’s diagnostic efficacy by simultaneously administering a muscarinic blocking agent, such as atropine. This could be done as follows: Two syringes are used. One contains saline; the other contains atropine, 0.4 mg, and edrophonium, 10 mg. Neither the patient nor the physician evaluating the response knows which of the two solutions is being injected. The nurse gives one syringe to the physician for injection. Three tenths of the volume is injected rapidly, and the response is evaluated during the next minute. If this is well tolerated, the remainder is injected and the response is evaluated. The contents of the second syringe are then injected in the same manner. Only after the patient and physician have committed themselves to an evaluation of the relative efficacy of the two syringes does the nurse identify which contained the edrophonium.
Several different parameters have been suggested for evaluating the edrophonium response. The objective improvements in ocular and lid motility have been of primary interest to the ophthalmologist. Glaser and co-workers224,225 used tonography to document the increased extraocular muscle tonus. They reported a mean 2-mmHg increase in intraocular pressure within 1 minute of injecting 10 mg edrophonium in 15 myasthenic patients. All 15 had unilateral, but not always bilateral, responses. The maximum elevation was 5 mmHg. The response could occur in less than 10 seconds, but the mean response interval was approximately 20 seconds226 to 35 seconds.227 Although there were no false-positive results in the 10 control patients in Glaser and co-workers’ study, false-positive results can occur.227,228 Wray and Pavan-Langston228 found that the intraocular pressure increase was greater in myasthenic patients without ophthalmoplegia than in those with ophthalmoplegia.
Kornbleuth and colleagues229 used electromyographic recordings of the extraocular muscles when testing with edrophonium. In their small series, only patients with myasthenia gravis showed an increase in electrical activity. Campbell and associates227 used electronystagmography to study the effect of edrophonium on the fatigue induced by optokinetic nystagmus. A beneficial response occurred in 50% of 17 myasthenia gravis patients given edrophonium injections. There were no false-positive reactions among 18 normal subjects. However, these authors found the test to be of only limited value because of the marked variations in amplitude and rate of optokinetic nystagmus among subjects.
Edrophonium increases the peak saccadic velocity in myasthenic patients but decreases it in normal controls.230 The use of topical cholinesterase inhibitor drops in the treatment of ocular myasthenia gravis has been suggested,231 but their value is not well documented.
Cholinesterase inhibitors are also used for their nicotinic effects in the treatment of pediculosis of the lashes. The cholinesterase inhibitors are pesticides and kill by producing respiratory arrest. Cholinesterase inhibitors used primarily for their intraocular effects are relatively ineffective pesticides. Physostigmine 0.1% in peanut oil and 0.25% ointment kill the adult form of the crab louse (Phithirus pubis), but the nits are more resistant and must be removed with forceps.232,233 Malathion is more effective and relatively safe because it is rapidly inactivated by human serum carboxylesterases. Within 5 minutes of exposure, 100% of lice are killed, and after 10 minutes of exposure 96% of eggs will not hatch. Two problems encountered when using malathion around the eyes are that commercial preparations contain a high (e.g., 78%) alcohol content and the drug gives off a foul-smelling sulfhydryl degradation product.
Demodex folliculorum is another pest infesting the lids. It is quite resistant to ophthalmic preparations. It can survive 3 or more days in 0.125% echothiophate, 0.25% demecarium, or 0.1% diisopropyl fluorophosphate.234
Nicotinic Antagonists
Two classes of nicotinic blocking agents exist: nondepolarizing antagonists (e.g., curare) and depolarizing antagonists (e.g., succinyldicholine).
NONDEPOLARIZING ANTAGONISTS. Neuromuscular Blockade. Duke-Elder and Duke-Elder235 showed that acetylcholine and nicotine caused contraction of dog extraocular muscles and that curare, but not atropine, could block these responses. Muscarine was subsequently shown to have a very weak contractile effect that could be blocked by atropine.236 Until the work of Katz and Eakins,237 it was believed that the ocular muscles were more sensitive to the blocking effects of nondepolarizing agents, such as curare and gallamine, than were the peripheral skeletal muscles.238 Katz and Eakins found that the reverse seemed true (e.g., a systemic dose of curare that paralyzed the cat anterior tibial muscle produced only a 50% block of the twitch response of the superior rectus). Their work was supported by Sanghvi and Smith.236 Systemic administration of nondepolarizing relaxants, such as alcuronium,239 atracurium,240,241 fazadinium,242 metocurine, and pancuronium,243 is associated with a lowering of intraocular pressure. Some of these have a rapid onset and short duration of action, which makes them useful as induction agents for intubation. These drugs also differ in their cardiovascular effects. For example, alcuronium and curare lower arterial blood pressure, whereas pancuronium increases blood pressure and heart rate.244
An effective neuromuscular block with atracurium or vecuronium lasts approximately 30 minutes, making these agents intermediate in their duration of action. Atracurium, unlike the shorter-acting depolarizing antagonist succinyldicholine, undergoes rapid ester hydrolysis at physiologic pH independent of serum butyrylcholinesterase activity. Large doses of atracurium have been suggested as a substitute for succinyldicholine induction because the former does not produce extraocular muscle contraction.245,246 Atracurium 0.5 mg/kg did not produce a significant rise in intraocular pressure in subjects under steady-state nitrous oxide general anesthesia, but succinyldicholine 1 mg/kg did, from a mean baseline of 5.6 mmHg to 13.2 mmHg. Vecuronium seems to act in a manner similar to that of atracurium.247,248
Mivacurium, at a dose of 0.15 mg/kg, has a slower onset of action (approximately 2.5 minutes) than succinyldicholine. At this dose, mivacurium’s striated muscle block lasts approximately 16 minutes. Mivacurium, unlike atracurium and vecuronium, is inactivated primarily by butyrylcholinesterase.
Traction on the extraocular muscles during general anesthesia produces slowing of the heart. As stated earlier, pancuronium produces a tachycardia, which is protective. Strabismic children under general anesthesia who received pancuronium had heart rates that dropped from 120 ± 17 beats per minute to 94 ± 25 beats per minute.249 Patients receiving atracurium had pulse rates that fell from 86 ± 20 beats per minute to 34 ± 22 beats per minute, and 40% developed an additional arrhythmia besides the bradycardia. From a cardiac viewpoint, pancuronium has advantages in strabismus surgery.
Ganglionic Blockade. Transmission in sympathetic ganglia (e.g., superior cervical ganglion) and parasympathetic ganglia (e.g., ciliary ganglion) is primarily nicotinic. Several investigators have looked at ocular effects that might be related to partial blockade at this level. Gallamine250 and pancuronium251 administration have been associated with a lowering of intraocular pressure. This may have been due, in part, to a reduction in systemic blood pressure. Intubation for general anesthesia produced tracheal irritation and a reflex increase in blood pressure. When throats were not anesthetized with topical anesthesia, the intraocular pressures 5 minutes after gallamine injection remained at baseline; when throats were anesthetized, the intraocular pressures were below baseline. The reductions in intraocular pressures produced by pancuronium were significant but were not dose related with the use of 0.01 to 0.08 mg/kg. Trimetaphan, a short-acting ganglionic blocking agent, administered intravenously produced reductions in intraocular pressure from preoperative values of 13 to 17 mmHg to intraoperative values of 3 to 4 mmHg as the systolic blood pressure fell to 60 mmHg.252
Hexamethonium and pentamethonium, 2 mg/kg, given intramuscularly to conscious subjects caused a mean maximum fall in intraocular pressure of 9 mmHg.253 The decline was present within 30 minutes of injection and persisted for 3 hours. This effect was attributed to a blockade of sympathetic ganglia and a resultant lowering of systemic blood pressure. Barnett253 treated hypertensive patients for 9 to 11 weeks with these drugs. He reported an improvement in the hypertensive retinopathy. However, these drugs produced an orthostatic hypotension that was disabling, which limited their use.
Retrobulbar tetraethylammonium injection in human subjects has produced mydriasis.254 The pupil remained dilated longer than 6 hours and did not constrict after topical physostigmine. Ciliary ganglion blockade also produced cycloplegia.255,256 Decreased tearing has been attributed to blockade of the sphenopalatine ganglion after systemic administration of tetraethylammonium.255
Based on the assumption that sympathetic superior cervical and stellate ganglia fibers innervate and constrict the retinal arterioles, ganglionic blocking agents have been used in the treatment of central retinal artery occlusions, in the hope that the vessels would dilate and the embolus pass to a more distal arterial branch. However, there is little evidence that they are beneficial, and it can be argued that they may further reduce the perfusion blood pressure to the eye.
Botulinum Toxin. Botulinum toxins are produced by the anaerobic bacteria Clostridia botulinum. Each of eight different strains produces an immunologically distinct form of the toxin: A, B, C1, C2, D, E, F, and G. Four, A, B, D, and E, have been shown to block cholinergic function. They bind to unmyelinated cholinergic nerve terminals and prevent the release of quantal acetylcholine.257,258 Death occurs by paralysis of the respiratory muscles; doses as low as 1 ng per kilogram body weight can be lethal in humans.259
Botulinum toxins are zinc-dependent proteases that attack proteins linking the acetylcholine-containing vesicles to the synaptic axon membrane. Calcium is no longer able to trigger a functional release of the transmitter.28 The structure of botulinum A toxin has been fully sequenced.260 Type A botulinum toxin attacks a soluble 25-kd cytoplasmic attachment protein, SNAP-25,29,261,262 abolishing quantal (i.e., functional) acetylcholine exocytosis; some nonquantal acetylcholine release does persist. The toxin does not block the neuron’s action potential, the synthesis or storage of acetylcholine, the vesicle number, or calcium entry.263 The initial binding site of botulinum A toxin is a sialic acid-containing site on the external neuron surface; this attachment can be antagonized by lectins.264 Once inside the neuron, botulinum toxin can no longer be neutralized by antitoxins.
Type A botulinum toxin is more effective at nicotinic neuromuscular junctions than at nicotinic ganglion synapses or parasympathetic neuromuscular junctions. It has been used to treat essential blepharospasm, blepharospasm associated with facial dyskinesias, hemifacial spasm, nonparalytic and thyroid ophthalmopathy-induced strabismus, paralytic strabismus, nystagmus, corneal exposure, and entropion.265,266,267,268,269,270,271,272,273,274,275,276,277,278
The toxin is injected into the muscle to be rendered paretic or subcutaneously in the adjacent area. The effect is dose dependent. Multiple small doses (e.g., 0.001 to 0.003 μg; 2.5 to 7.5 U) or fewer but larger doses may be given. The onset of effect is within 48 hours, and the maximum effect may take 5 to 6 days to develop. The results are usually transient and average 2 to 3 months in duration.
Histologic examination of orbicularis muscle fibers after prolonged treatment of blepharospasm failed to show morphologic changes. However, the motor neurons had many unmyelinated axon sprouts, most of which ended blindly; those ending on muscle fibers were able to elicit new and multiple motor end plates at previously nonsynaptic sites.279,280
In general, botulinum A treatment is of little value in strabismus in which the eye muscle movements are restricted, and it is of less value than traditional surgery in comitant deviations.281 Retrobulbar injections, used to treat nystagmus and oscillopsia, may be of temporary value depending, in part, on the amount and duration of ptosis produced.282,283,284,285 Complications occur from local spread of toxin to adjacent muscle fibers and from an inability to accurately predict the magnitude of the effect. Ptosis is one of the most common complications caused by local spread of toxin.286 Ptosis, partial or complete, can at times be of therapeutic value (e.g., before recovery from Bell’s palsy).287,288,289 Deliberate ptosis can be achieved by injecting botulinum A toxin along the superior orbital roof, approximately 25 mm behind the superior orbital rim.
Clinical signs of systemic toxicity have not been reported, but there is evidence of subtle systemic effects (e.g., abnormal electromyographic recordings in arm muscle fibers of patients receiving 0.005 μg of toxin).290 Neurologists treat dystonias with relatively large doses of botulinum A toxin; these may produce only subtle distant electromyographic changes but are likely to elicit neutralizing antibodies that limit the toxin’s usefulness.291,292,293 A distinction should be made between non-neutralizing antibodies, which may have no clinical significance, and neutralizing antibodies.294
DEPOLARIZING ANTAGONISTS. Extraocular Muscle Effects. The depolarizing antagonists, such as succinyldicholine, decamethonium, and hexacarbacholine (Fig. 2), paralyze striated voluntary muscles throughout the human body but have a different effect on the extraocular muscles. The extraocular muscles respond with a sustained contraction. This was first noted by Hofmann and Holzer,295 who reported that the contraction of the extraocular muscles caused an increase in human intraocular pressure. This observation was supported by Lincoff and co-workers.296,297 The maximum intraocular pressure reported is 55 mmHg.298
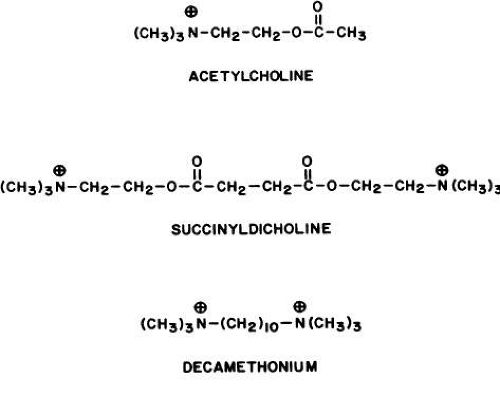 Fig. 2. Succinyldicholine and decamethonium mimic the structure of acetylcholine to some extent. Succinyldicholine is two molecules of acetylcholine joined at the acetate ends. It and decamethonium will cause a sustained contraction of those extraocular muscle fibers that are multiply innervated. |
The shortening effect and increased muscle tension created by succinyldicholine have been measured in humans.299 The drug always produced contraction in the horizontal and vertical recti muscles. The results in the oblique muscles were variable.
The explanation is that there are two types of extraocular muscle fibers. In the center of each muscle, typical striated muscle fibers, with a single myoneural junction (en plaque fibers), are most numerous. These are paralyzed by succinyldicholine. However, the surface layers contain fibers with multiple myoneural junctions. These are the “en grappe” fibers. Their innervation has the appearance of a bunch of grapes. It is these fibers that respond to succinyldicholine with a sustained contraction.300,301,302 The strength of contraction depends on the dose of succinyldicholine and the number of multiply innervated fibers. In sheep superior oblique muscle, succinyldicholine produces only 7% of the force generated by maximum electrical stimulation of the entire muscle.303 In cats, up to one third of the whole muscle force has been generated by succinyldicholine.304 Succinyldicholine causes a partial reduction in the resting membrane potential and a localized contraction in the area around each myoneural junction.305 Because these junctions are found along the entire length of the en grappe fiber, most of each fiber is depolarized and shortened.306 However, unlike en plaque fibers, which are capable of generating action potentials, the en grappe fibers are believed to respond to both neuronal impulses and succinyldicholine with slow, graded contractions (i.e., they are unable to conduct action potentials).307 This may explain why Kornbleuth and colleagues229 found a dissociation between ocular muscle electrical activity and the elevation of intraocular pressure. They performed simultaneous tonography and electromyography on subjects given succinyldicholine. They expected to find that the increase in intraocular pressure was proportional to the degree of extraocular muscle electrical activity. Electromyography was performed on antagonistic vertical or horizontal recti of the same eye in subjects receiving 5, 10, 15, or 20 mg succinyldicholine. These doses were too small to produce apnea. A 2- to 4-mmHg increase in intraocular pressure occurred in patients under general anesthesia, and a 5- to 14-mmHg increase occurred in conscious volunteers. The duration, but not the magnitude, of the ocular pressure elevation appeared to be dose related. The rise in intraocular pressure elevation occurred just as the electrical activity of the extraocular muscle was reduced. The inability of the en grappe fibers to conduct action potentials may explain this dissociation.
Curare, but not atropine, blocked the sustained contraction produced by succinyldicholine in animal studies.308 Katz and Eakins237 found evidence that the en plaque fiber response to depolarizing antagonists was quantitatively different for eye muscles and skeletal muscles (e.g., the dose of succinyldicholine needed to depress the twitch response of the superior rectus muscle was greater than that needed to block skeletal muscles). This difference was less when decamethonium was used.
Each of the six human extraocular muscles contains multiply innervated and singly innervated fibers in about the same proportions: in the periphery, 92% ± 10% are multiply innervated, and in the center, 33% ± 12% are multiply innervated.309 The contractile effect of succinyldicholine on the en grappe fibers is generally considered undesirable. However, Hannington-Kiff310 suggested that the anesthesiologist use the resultant change in interlimbal distance to monitor the action of succinyldicholine. The interlimbal distance in 35 patients under general anesthesia before succinyldicholine injection was 52.9 mm ± 4.3 mm. Ninety seconds after injection, this distance had decreased to 49 mm ± 5.5 mm. One minute after the return of spontaneous respirations, the interlimbal distance was near baseline values, 51.4 mm ± 3.3 mm. The duration of succinyldicholine stimulation of en grappe fibers and the duration of succinyldicholine paralysis of en plaque fibers appeared similar. Taylor and associates311 also found that the intraocular pressure returned to baseline with the onset of spontaneous respirations. Pandey and co-workers312 studied the time course of the ocular pressure elevation in 34 patients given succinyldicholine in a dose of 1.5 to 2 mg/kg. An increase in intraocular pressure was present in less than 1 minute, peaked in 2 to 4 minutes, and returned to baseline in 6 minutes.
Succinyldicholine stimulation of the extraocular muscles results in a variable amount of enophthalmos; as much as 3.25 mm has been reported.313 The conjunctival vessels dilate,314 and the forced-duction test may suggest a mechanical restriction for up to 20 minutes.315
Mindel and colleagues316 theorized that the basic deviation of the eyes was produced by the balance of forces exerted by the multiply innervated muscle fibers. They used intravenous succinyldicholine to stimulate these fibers and to remove the influence of the singly innervated fibers, which were paralyzed. Under general anesthesia, esotropic patients became esotropic when 2 mg/kg was injected. Exotropic patients became exotropic.317 Despite this qualitative success, the measured amount of drug-induced ocular deviation did not agree closely with that in the conscious patient. These authors believed that this incongruity was due to A and V pattern effects, that is, in conscious patients the eyes could be maintained in the primary position by fixation, whereas in anesthetized patients the eyes could move vertically as well as horizontally. Lingua and associates318 have reported similar findings. In addition, they found a correlation between the ocular position induced by succinyldicholine immediately after strabismus surgery and the 1-week postoperative result.
Intraocular Muscle Effects. Abramson319 used ultrasound to measure anterior chamber depth and lens thickness in human subjects given 1 mg/kg succinyldicholine intravenously. Succinyldicholine caused a rapid increase in anterior chamber depth and a decrease in lens thickness. This effect was present within 20 seconds, was maximal in 45 to 210 seconds, and disappeared in 3 to 5 minutes. Abramson attributed this effect to a relaxation of accommodation. However, there was no change in the diameter of the pupil. It is unlikely that succinyldicholine would decrease the activity of only one of the two parasympathetic structures innervated by the ciliary ganglion.
Side Effects. Succinyldicholine may produce a bradycardia. This is usually associated with multiple injections. However, asystole has been reported after a single dose.320 Succinyldicholine and, to a lesser degree, its first hydrolysis product, succinylmonocholine, are responsible for this cardiac effect.321
Succinyldicholine is inactivated by esterase hydrolysis. The apnea produced by respiratory muscle paralysis may be prolonged in patients with genetically322 or acquired reduced serum cholinesterase activities.323,324 Chronic systemic absorption from topical cholinesterase inhibitor eyedrops can result in such an effect.325 In one report, a reduction of serum cholinesterase activity 62% below normal from echothiophate eyedrops permitted endotracheal intubation using a total of only 9.5 mg succinyldicholine.326 Decamethonium, because it is excreted unmetabolized, is not dependent on the level of serum cholinesterase activity for termination of its effect.
There are at least four alleles, at a single locus, that control the type of serum cholinesterase made by the liver: normal, dibucaine resistant, fluoride resistant, and silent genes. Approximately 1 in 4000 in the population will develop prolonged apnea after succinyldicholine injection. This is the incidence of a person having both genes abnormal. Approximately 1 in 400 in the population will be a normal dibucaine-resistant heterozygote and will develop a shorter but still significantly prolonged apnea. Other factors reducing serum cholinesterase activity are hepatic disease,327 malnutrition, anemia, severe dehydration, late pregnancy,328 and systemic absorption of cholinesterase inhibitors. In one study, 5% of a general surgical population had decreased serum cholinesterase activity.329
The sustained contraction of the extraocular muscles after succinyldicholine injection poses a threat if there is a preexisting perforating laceration or if an incision is to be made into the eye (e.g., cataract extraction). Expulsion of the intraocular contents can result from the steady squeezing of the eye muscles. Lincoff and colleagues297 mentioned three personal communications linking the use of succinyldicholine to this complication. There are several ways it can be avoided: After succinyldicholine injection, the intraocular pressure is monitored with a tonometer until it returns to normal. Use of depolarizing nicotinic antagonists is avoided. Alternatively, a pharmacologic antagonist can be used to prevent the effect. With regard to this last suggestion, cholinesterase inhibitors are not of value. Although they reverse the action of nondepolarizing muscle relaxants (e.g., curare), they prolong and enhance the action of depolarizing drugs.330 Theoretically, pretreatment with small doses of curare or succinyldicholine itself might prevent succinyldicholine-induced ocular muscle contraction. However, the results are conflicting.331,332 Macri and Grimes333 found that prior curare injection prevented ocular hypertension in cats given succinyldicholine. Because both drugs competed for the same receptor, multiple doses of succinyldicholine overcame the curare effect. Miller and associates334 found that 1 mg/kg succinyldicholine produced a mean intraocular pressure increase of 8.5 mmHg in 10 control subjects. Parenteral gallamine, 20 mg, or curare, 3 mg, administered 3 minutes before succinyldicholine, prevented this effect in both glaucomatous and normal subjects. However, when Meyers and co-workers335 studied the value of curare, 0.09 mg/kg, or gallamine, 0.3 mg/kg, given 3 minutes before 1 to 1.5 mg/kg succinyldicholine, they found that ocular pressure elevations still occurred (e.g., in 10 patients with a mean intraocular pressure of 13 ± 1 mmHg, curare did not prevent an elevation to 24 ± 1.3 mmHg). Pancuronium, 0.14 mg/kg, also was ineffective.336 Katz and colleagues337 found that hexafluorenium, which is both a nondepolarizing (curarelike) blocking agent and a plasma cholinesterase inhibitor, in doses of 0.4 mg/kg, prevented the rise in intraocular pressure from succinyldicholine, 0.3 mg/kg. Hexafluorenium was effective only if it was injected before succinyldicholine, not after it.
Perhaps the preceding inconsistencies result from the multiplicity of factors that can affect the intraocular pressure (e.g., depth of anesthesia, serum O2 and CO2 levels, venous pressure, and tracheal stimulation). Barbiturates, given to induce anesthesia, can reduce muscle endplate currents.338 When thiopental was injected 2 minutes before succinyldicholine, 1 mg/kg, there was no significant increase in ocular pressure.339 Macri and Grimes333 disinserted all the extraocular muscles in cats. This reduced, but did not prevent, an increase in intraocular pressure after succinyldicholine. Taylor and associates311 reported that in 25 of 29 patients, the blood pressures 1 minute after succinyldicholine injection were elevated. These may have contributed to the short-term elevations in ocular pressure.
The blood pressure elevations could be because succinyldicholine raises blood catecholamine levels. Succinyldicholine, but not curare, increased plasma norepinephrine levels immediately after injection.340 Succinyldicholine, 1 mg/kg, produced a peak norepinephrine elevation 3 minutes after injection, raising levels from a mean of 301 pg/mL at baseline to 647 pg/mL. The increase had disappeared by 10 minutes. The elevation in epinephrine levels was less marked and became insignificant by 2 minutes.
Wynands and Crowell341 found that 50% of patients given succinyldicholine had a rise in intraocular pressure. In 3 of 18 subjects, the elevation was more than 10 mmHg. However, endotracheal intubation alone produced a rise of more than 10 mmHg in 10 of 23 subjects. Lewallen and Hicks342 and Craythorne and co-workers298 reported conflicting results in subjects without intubation. The former found that injection of 40 mg succinyldicholine did not raise human intraocular pressure; the latter found that continuous infusion of 0.1% to 0.2% succinyldicholine did.
A most intriguing study was that of 15 patients having uniocular enucleations.343 These subjects were anesthetized with 3 to 4 mg/kg thiopental intravenously and maintained with halothane or isoflurane and nitrous oxide/oxygen. No premedications were used. All six extraocular muscles were severed from the eye to be enucleated. Intraocular pressures were then obtained bilaterally. Succinyldicholine 1.5 mg/kg was administered and the intraocular pressures were measured every 30 seconds for 5 minutes. The systolic and diastolic blood pressures were not significantly different before and 90 seconds after succinyldicholine administration, but the intraocular pressures were significantly and maximally elevated bilaterally at this time. Surprisingly, there was no significant difference in the mean ± SE intraocular pressure rise between those eyes with severed muscles and those eyes with muscle insertions intact. The presuccinyldicholine injection intraocular pressures of the normal and muscle-severed eyes were, respectively, 15.1 ± 1 mmHg and 16.1 ± 1 mmHg. At 90 seconds after succinyldicholine injection, the respective intraocular pressures were 25.2 ± 1.6 mmHg and 24.7 ± 1.8 mmHg. Perhaps an increase in venous blood pressure produced these bilateral results, but the apparent lack of an extraocular muscle effect is inexplicable.
MUSCARINIC DRUGS
Direct-Acting Muscarinic Agonists
GENERAL CONSIDERATIONS. The ocular effects of this class of drugs include reduction in intraocular pressure; stimulation of the iris sphincter, producing miosis; and stimulation of the ciliary muscle, producing accommodation. Examples of direct-acting muscarinic drugs are muscarine, pilocarpine, aceclidine (3-acetoxyquinuclidine), arecoline, and acetyl β-methylcholine (methacholine, Mecholyl) (Figs. 3, 4, 5). In high concentration, pilocarpine also stimulates choline acetyltransferase synthesis of acetylcholine,344 but this is probably of little clinical significance.345
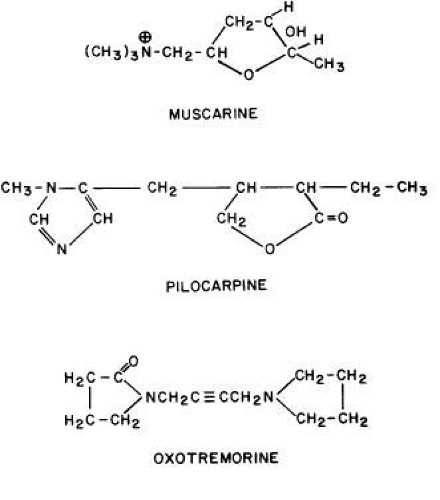 Fig. 3. Naturally occurring direct-acting muscarinic agonists. |
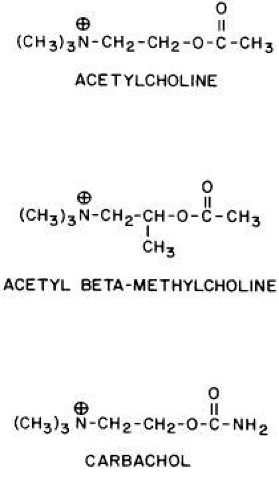 Fig. 4. Synthetic congeners of acetylcholine. Acetyl β-methylcholine is a muscarinic agonist. Carbachol is both a nicotinic and a muscarinic agonist. |
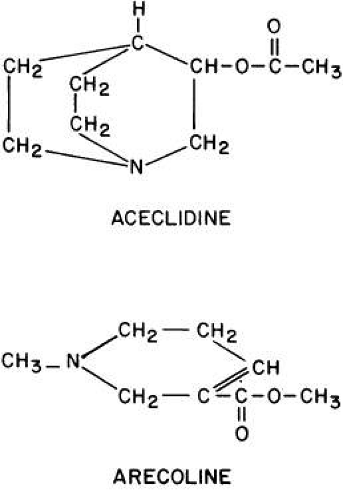 Fig. 5. The direct-acting muscarinic agonists aceclidine and arecoline are, like pilocarpine, tertiary amines. They penetrate the corneal epithelium more readily than do the positively charged quaternary amine drugs. |
Isopilocarpine is a naturally occurring stereoisomer of pilocarpine present in very small amounts in most preparations of the drug.346 Isopilocarpine has one tenth the binding affinity of pilocarpine for bovine ciliary muscle and has very little pharmacologic activity.
Carbachol (carbamylcholine) is both a direct-acting muscarinic agonist and a direct-acting nicotinic agonist. In addition, it has indirect agonist activities (i.e., it is a cholinesterase inhibitor). This last action results from the NH2 substitution on the acetyl group of carbachol. It markedly reduces the molecule’s susceptibility to hydrolysis by acetylcholinesterase but does not prevent carbachol from competing with acetylcholine for the anionic and esteradic sites of acetylcholinesterase. This interference with acetylcholine hydrolysis results in a further increase in cholinergic activity. Aceclidine, too, is slightly cholinesterase resistant and has weak anticholinesterase activity.
Pilocarpine’s structure is quite different from that of acetylcholine. Either of the nitrogen atoms in the imidazole ring can become positively charged and bind to the anionic receptor site. Pilocarpine does not have the classic muscarinic agonist structure of acetylcholine (N-C-C-O-C-C). However, pilocarpine’s interatomic spacing (N-C-C-C-C) may mimic it.347 Evidence exists that the intrinsic muscarinic activity of pilocarpine is less than that of acetylcholine and carbachol.348,349 However, additional factors play a role in determining clinical efficacy. These are primarily pharmacokinetic. For example, acetylcholine and acetyl β-methylcholine are hydrolyzed by tissue esterases and may not reach the site at which their action is desired. Inhibiting tissue cholinesterases increases the efficacy of both drugs. Arecoline, aceclidine, and pilocarpine are tertiary amines and can penetrate the corneal epithelium more readily than the positively charged quaternary amines such as acetylcholine, carbachol, and acetyl β-methylcholine. This correlates with the lipid solubilities of these drugs. As measured with toluene: pH 6.5 buffer, the partition coefficient of arecoline is 0.1; pilocarpine, 0.033; aceclidine, 0.002; and carbachol, <0.0001.350 The pHs of eyedrops and tears alter the proportions of nonionized drug (e.g., the ability of arecoline and oxotremorine to reverse mydriasis increases as a function of the pHs of their solutions).351 Drugs that are ionized at all pHs, such as carbachol, are concentration dependent, not pH dependent. The poor corneal penetration of carbachol resulted in Clarke352 finding marked variability in its ability to lower intraocular pressure. O’Brien and Swan353 performed a corneal massage through the lids of human subjects. This allowed a 0.09% carbachol solution to be as effective a miotic as a 1.5% to 4.5% carbachol solution. Topical anesthetics and surfactants also aided penetration (e.g., the addition of 0.03% benzalkonium to 1.5% carbachol produced significant accommodation for 48 hours).
Accurate estimates of corneal penetration by pilocarpine have been impaired by the use of tritiated molecules. Asseff and co-workers354 gave anesthetized monkeys eyedrops of 1% to 8% tritiated pilocarpine. They reported approximately 3% corneal penetration at 5 minutes. Chrai and Robinson,355 studying rabbits, found similar levels after applying unpurified tritiated pilocarpine but only 0.13% corneal penetration at 5 minutes after applying purified material. Maximum penetration occurred at 20 minutes and was 0.18%. At 2 hours, the aqueous humor concentration was 0.03% of the administered dose. These authors attributed the previous higher values to tritium exchange between pilocarpine and its solvent during storage. Penetration of the tritiated solvent resulted in falsely high estimates of corneal penetration. Krohn and Breitfeller356 applied two drops of 2% pilocarpine HCl (approximately 2000 μg) to corneas of patients under general anesthesia; the pilocarpine concentration in the aqueous humor was not more than 5 μg/mL.
Pilocarpine can be administered from continuous-release membranes (Ocusert) made of polymerized ethylenevinylacetate. Ideally, the low-dosage form (Ocusert P20) releases 20 μg/h and the high-dosage form (Ocusert P40) releases 40 μg/h. However, Armaly and Rao357 found that the in vitro release rates from membranes supposedly dispensing 20 μg/h, 50 μg/h, and 80 μg/h were two and one half to three times their projected rates during the first 7 hours of use. Between 7 and 24 hours, the release rates remained slightly elevated, but within the next 24 hours they fell to slightly below their projected levels and continued to decline. After 3 to 4 days, the release rates were 78%, 88%, and 81% of those expected. These results may explain why the glaucoma of two of Armaly and Rao’s patients was controlled for the first 2 days of membrane use but not thereafter.
Pilocarpine has also been applied as a polymer emulsion358,359 and as a gel. Both provide prolonged drug delivery to the eye and, compared with drops, longer duration of action. The gel is available commercially as 4% pilocarpine HCl in a viscous aqueous acrylic gel vehicle; this concentration of pilocarpine approaches the binding limit of the polymer used to make the gel. A single daily application of the gel has a hypotensive effect for 24 hours. For the first 18 hours, this effect is indistinguishable from that elicited by 4% pilocarpine eyedrops given four times a day in terms of both pressure and pupillary diameter.360 For hours 18 to 24, the drops are more effective.361
Pathology can alter pharmacokinetics and drug response. Patients with such diverse pathology as atopy and trisomy 21 are said to be more sensitive to muscarinic agonists and antagonists.362,363
Patients with intraocular inflammation inactivate pilocarpine. Schonberg and Ellis364 found that the primary aqueous humor of humans and rabbits did not inactivate pilocarpine, but serum and secondary aqueous did. Ten percent of 500-μg pilocarpine was inactivated by 200 μL of secondary aqueous humor in 1 hour. Inactivation could be prevented by heating serum to 60°C for 10 hours, by dialysis, or by the addition of some (e.g., penicillamine and ethylenediaminetetraacetic acid), but not all, chelating agents. Inactivation could not be inhibited by anticholinesterase drugs. Rabbit serum was more active against pilocarpine than human serum. Only at high pilocarpine concentrations were human ocular tissues (cornea, iris-ciliary body, lens, retina, and choroid) more active than the corresponding rabbit ocular tissues.365 Lee and colleagues366 found that pigmented rabbit corneas inactivated pilocarpine much more effectively than did albino rabbit corneas.
OCULAR PHARMACOLOGY Iris Sphincter Muscle. ANIMAL STUDIES. Pilocarpine has less intrinsic muscarinic activity than the endogenous transmitter acetylcholine. As a result, Swan and Gehrsitz367 found that rabbits given topical 0.25% physostigmine or 0.1% DFP had less miosis if 4% pilocarpine was previously administered. If pilocarpine were injected into the cornea after maximum DFP miosis, a transient dilatation occurred. These results were consistent with pilocarpine-acetylcholine competition for the same receptors.
Miosis in mammals is associated with M3 receptor activation and increased phospholipase C activity.368,369 Feedback inhibitory prejunctional muscarinic receptors on sympathetic neurons of the iris dilator muscle seem to be of a different subtype.199 In rabbits, these prejunctional receptors respond to methacholine, oxotremorine, muscarine, and carbachol by inhibiting norepinephrine release; atropine can block this effect.370
Denervation supersensitivity of the rat iris sphincter is not associated with a change in muscle fiber resting potential but is associated with increased sensitivity to calcium.371,372 Thus, supersensitivity could be due to an increased release of intracellularly bound calcium and/or an increased influx of extracellular calcium.
Bito and Banks373 administered DFP drops chronically to monkeys and then discontinued the drug. They observed several interesting pupillary phenomena, one of which was a diminished response to direct-acting muscarinic agonists during the recovery period. For at least 3 days after discontinuing DFP, the pupils responded to light but did not respond to carbachol or pilocarpine. Similar results were obtained in the cat.374 These results may have been produced from an acquired subsensitivity of the sphincter muscarinic receptors (i.e., a subsensitivity produced by the prior high acetylcholine concentrations). Another possibility is that the greater intrinsic agonist activity of acetylcholine, compared with that of pilocarpine and carbachol, became manifest. Other data from this laboratory,375 confirmed by Claesson and Barany,376 suggested that an alteration of muscarinic receptor sensitivity had occurred. Exposure of feline eyes in vivo to continuous light resulted in subsensitivity to pilocarpine, whereas light deprivation resulted in increased sensitivity. After 7 days in ambient light of 1200 lumens/m2, the irides became insensitive to pilocarpine. Surprisingly, subsensitivity to carbachol or acetyl β-methylcholine could not be demonstrated in vitro. Hemicholinium (which reduces acetylcholine synthesis) injected intravitreally in vivo resulted in increased sensitivity to administered muscarinic agonists. Barany377 reported that chronic administration of pilocarpine drops did not produce subsensitivity in experimental animals challenged with intravenous pilocarpine. However, after prolonged treatment in the form of continuous-release membranes, pilocarpine did produce subsensitivity.
Perhaps the explanation for the aforementioned phenomena is miosis from peptide transmitters. There is evidence that the neurotransmitter substance P, identified in trigeminal nerve fibers, produces miosis.378 Mandahl379 has suggested that, in the rabbit, the miosis elicited by cholinesterase inhibitors and pilocarpine is primarily the result of release of substance P from trigeminal nerve endings in the iris. According to this theory, continuous application of cholinesterase inhibitors and pilocarpine would deplete the stores of substance P, resulting in a reduced response, whereas the acetylcholine release by light-stimulated parasympathetic fibers would remain intact.
HUMAN STUDIES. Lowenstein and Loewenfeld380 and Newsome and Loewenfeld381 studied the iris using pupillography. Drops of 1% to 2% pilocarpine were given to volunteers without ocular disease. The onset of miosis, maximum miosis, and duration of miosis varied greatly from person to person. Maximum miosis occurred in 25 to 45 minutes, was maintained for 1 to 3 hours, and required 24 to 36 hours for full recovery. The pupillary reflex to light persisted unless miosis was maximal. However, during the recovery phase, the light response was markedly diminished. When submaximally effective concentrations of pilocarpine (e.g., 0.1%) were followed by submaximal concentrations of a cholinesterase inhibitor (e.g., 0.1% physostigmine), the results were additive.
Ogle and associates382 and Morgan and co-workers383 also used pupillography to study miosis in normal subjects. They reported latencies after 1% pilocarpine of 9.8 ± 0.9 minutes. The latencies for 0.75% carbachol and 0.25% physostigmine were 13.7 ± 2.2 minutes and 13.9 ± 1 minutes, respectively. The rates of miosis and amounts of maximum miosis were less for carbachol than for pilocarpine and physostigmine, yet all three drugs required about the same time to produce their maximum effect. The times required for recovery to 50% of the predrop pupil diameters were: carbachol, 164 ± 46 minutes; pilocarpine, 271 ± 109 minutes; and physostigmine, 456 ± 28 minutes. The latency of mydriasis from 0.5% tropicamide (Mydriacyl) was similar to the latency of miosis from pilocarpine. The changes in pupillary diameter due to light stimuli during the latency periods of both pilocarpine and tropicamide were similar; the amounts and maximum rates of pupil constriction after onset of action also were similar.
After the short-term application of various direct-acting muscarinic agonists, the response produced by the light reflex was reduced.384 This may have represented competition between residual exogenous agonists, with weaker intrinsic activity, and the endogenous agonist acetylcholine, with more potent intrinsic activity. The light reflex required approximately 24 hours to recover after 2 drops of 2% pilocarpine, 7 hours to recover after 10 drops of 0.2% arecoline hydrobromide, and 24 hours to recover after 10 drops of 0.2% aceclidine hydrochloride.
Ciliary Muscle. ANIMAL STUDIES. Tornqvist385,386,387 studied accommodation in monkey eyes. Intramuscular pilocarpine over a dose range of 0.2 to 2 mg/kg appeared to have equal affinity for iris and ciliary muscle. The lowest dose caused 2 diopters (D) or less of accommodation, and the highest dose always caused more than 12 D of accommodation. One monkey was given higher doses of pilocarpine, and the myopia paradoxically decreased: 2 mg/kg produced 20.3 D of accommodation, 5 mg/kg produced 11.7 D of accommodation, and 50 mg/kg produced 8 D of accommodation. Similarly, small doses of systemic carbachol, 0.005 to 0.1 mg/kg, produced increasing accommodation, but larger doses, 0.1 to 0.5 mg/kg, produced decreased accommodation. This phenomenon could not be reproduced by using high concentrations of topical muscarinic agonists. Retrobulbar anesthesia did not prevent this paradoxical effect, indicating that central nervous system and ganglionic effects were not the cause. Intravenous atropine prevented, or reversed, the paradoxical effect. A topical dose of pilocarpine that produced half the maximum accommodative effort was 100 times more than that needed to produce half the maximum miosis. Because systemically administered pilocarpine did not exhibit this difference, it was concluded that pharmacokinetic factors resulted in more drug reaching the iris than reached the ciliary muscle after topical application.
Barany388 studied subsensitivity of the monkey’s ciliary muscle after a single drop of topical carbachol given unilaterally. For approximately 1 week, the accommodative response in the treated eye was less than that in the untreated eye after systemic administration of pilocarpine, carbachol, or bethanechol. The reasons for this effect were not clear, but the author did not believe the answer was a reduction in the number of muscarinic receptors or a reduction in their binding characteristics.
In monkeys, the loss of accommodation that occurs with aging is not associated with a reduction in the number of ciliary muscle muscarinic receptors.389
The agonist aceclidine is less effective in producing accommodation than in increasing miosis and outflow facility. Studies in monkey eyes found that M3 receptors predominate in ciliary muscle, iris sphincter muscle and trabecular meshwork (i.e., the differences in pharmacologic responses could not be attributed to different muscarinic receptor subtypes).390,391
After ciliary ganglionectomy, monkey ciliary muscle supersensitivity to pilocarpine was associated with a 12% to 84% reduction in muscarinic receptors.392 The magnitude of this seemingly paradoxical finding is difficult to explain simply by attributing it to the loss of prejunctional muscarinic receptors on nerve endings.
HUMAN STUDIES. Ciliary body thickness was measured in volunteers by the use of ultrasound.393 Two hours after applying a drop of 2% pilocarpine, the increase in mean thickness was 0.06 mm. Anterior chamber depth was measured after single and multiple drops of 2% or 4% pilocarpine.394 All subjects showed a decrease in anterior chamber depth after a single drop of pilocarpine. This shallowing remained significant for the 6 hours that measurements were made. When compared with the untreated eye, a single drop of 2% pilocarpine produced a mean maximum shallowing of 0.09 mm that occurred 3 hours after instillation; 4% pilocarpine produced a mean maximum shallowing of 0.07 mm that occurred 4.5 hours after instillation. Pilocarpine (2%), every 6 hours, produced a mean maximum anterior chamber narrowing of 0.12 mm. This was not significantly different from the single-drop effect. One week after chronic therapy was discontinued, the mean depth of the treated eye was not significantly different from that of the control eye. Abramson and colleagues395 confirmed that the anterior chamber shallowing effect from 2% pilocarpine, as well as the increase in lens thickness, was similar after single-drop and chronic (10-day) therapy.
Hallden and Henricsson396 produced with-the-rule astigmatism in 10 young men by asymmetrically contracting the ciliary body. This was achieved by applying a cotton pledget containing 10% pilocarpine to the temporal bulbar conjunctiva for 3 minutes, creating 0.75- to 2 D myopia with an axis between 0° and 20°.
Intraocular Pressure. The site at which muscarinic drugs act to lower intraocular pressure is a subject of controversy. Their effect is believed to be local, although an occasional report raises the possibility of a systemic effect (e.g., Willetts397 reported that normotensive subjects given unilateral pilocarpine drops had a bilateral decrease in ocular pressure). However, subjects with a mean plasma level of 2.9 ng/mL, delivered by skin patches, did not have a significant reduction in intraocular pressure.398
The different mechanisms of action that have been proposed include a decrease in aqueous humor secretion; an increase in removal of aqueous humor by active transport; an increase in removal of aqueous humor by passive trabecular outflow (from increased iris sphincter tone, increased ciliary muscle tone, or decreased episcleral venous pressure); and an increase in removal of aqueous humor by way of nontrabecular routes.
AQUEOUS HUMOR SECRETION. The data from different laboratories are inconsistent. Becker399 found that pilocarpine drops increased rabbit aqueous humor formation in vivo. Green and colleagues400 reported that acetylcholine and pilocarpine produced an increase in rabbit aqueous humor secretion in vitro. Macri and Cevario401 administered pilocarpine intra-arterially to enucleated cat eyes and also found an increase in aqueous humor secretion; however, outflow facility was increased, producing an overall reduction in intraocular pressure. Berggren402,403 found the opposite results. He used in vitro thinning of rabbit ciliary processes as a measure of secretion. Pilocarpine 10-9 mol/L, physostigmine 10-7 mol/L, acetylcholine 10-5 mol/L, and carbachol 10-5 mol/L inhibited secretion. The therapeutically inactive stereoisomer of pilocarpine, isopilocarpine, did not block secretion. Atropine 10-7 mol/L alone had an inhibitory effect402 and also prevented the more potent pilocarpine effect.403 However, when intravenous pilocarpine, 2 mg/kg, was injected in vivo 5 minutes before the in vitro studies, there was no evidence of altered secretion.
Edwards and colleagues,404 using constant-pressure perfusion, measured the effect of unilateral topical 4% pilocarpine on monkeys under general anesthesia. Pilocarpine caused an increase in aqueous humor production. However, Walinder and Bill405 found in vervet monkeys that infusion of pilocarpine 10-4 mol/L into the anterior chamber produced a reduction in aqueous humor formation of approximately 1 μL/min; atropine infusion 3 × 10-5 mol/L abolished the pilocarpine effect.
Barsam406 found that the hypotensive effect of pilocarpine lasted longer than the duration of increased outflow facility in human subjects. He believed that pilocarpine was producing a reduction in aqueous humor secretion. However, fluorophotometry of normotensive eyes has provided evidence that pilocarpine has only a minor effect on aqueous humor formation, which is to increase secretion by approximately 14%.407 This may or may not be true for the glaucomatous eye as well.
Muscarinic agonists might alter aqueous humor secretion by altering ciliary blood flow. Stjernschantz and Bill408 studied cholinergic control of uveal blood flow in anesthetized animals. Stimulation of the intracranial third nerve altered the blood flow to the iris and ciliary body but not to the retina and choroid. In the cat, the ciliary body blood flow was increased; in the rabbit, it was decreased; and in the monkey there was no statistically significant effect. In all three species, the iris blood flow was decreased by third-nerve stimulation. Intravenous atropine blocked this iris effect in all three species and prevented the increased ciliary body blood flow in cats.
AQUEOUS HUMOR ACTIVE TRANSPORT. Large vacuoles have been observed in the endothelium of the inner wall of Schlemm’s canal. It has been suggested that they represent the result of active transport of aqueous humor. Holmberg and Barany409 found that monkey eyes treated with pilocarpine had a reduced number of vacuoles. Lutjen-Drecoll410 placed 20 μg pilocarpine hydrochloride into the anterior chambers of monkeys and then studied the eyes with electron microscopy. Large vacuoles were rarely encountered. Grierson and associates411 studied the enucleated eyes of eight patients with melanomas. Pilocarpine drops were given before their removal. These were compared with 11 control eyes but not in a masked manner. There were more than twice the number of large vacuoles in the pilocarpine-treated eyes. However, the authors’ results from baboon eyes led them to conclude that the increased vacuolization was not due to a direct effect of pilocarpine on the endothelium of Schlemm’s canal. Instead, they attributed the vacuoles to a secondary effect from increased trabecular flow.412
INCREASE IN PASSIVE AQUEOUS HUMOR OUTFLOW. Most investigations have dealt with this mechanism of muscarinic action. Edwards and co-workers404 found that topical 4% pilocarpine caused more than a doubling in monkey eye outflow facility, from 0.41 ± 0.04 μL/min/mmHg to 1.09 ± 0.14 μL/min/mmHg. Atropine alone had no effect. Barany413 found that pilocarpine, administered topically or injected into the anterior chamber, increased vervet monkey facility of outflow. Two components appeared to be present, because atropine, injected intravenously or intraocularly, reversed part of the increase in 3 to 5 minutes and the remainder in approximately 30 minutes. Lutjen-Drecoll410 studied electron micrographs of monkey eyes and found that the trabecular pore area in contact with the inner wall of Schlemm’s canal correlated, after pilocarpine administration, with the outflow facility.
In humans, the hypotensive effect of pilocarpine is usually, but not always, associated with an increase in outflow facility. Outflow facility, accommodation, and pilocarpine-induced accommodation decrease with age. However, the pilocarpine-induced increase in outflow facility and reduction in intraocular pressure were found not to decrease with age.414 Pilocarpine, using gonioscopy to evaluate the drug’s effect, seems to produce a bowing inward of the trabecular meshwork toward the anterior chamber and away from the canal of Schlemm.415
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


