33 Cancer of the Temporal Bone
Cancer of the temporal bone is a rare but important pathology. Without a high index of suspicion in the appropriate circumstances, the diagnosis can be missed.
Its presentation can be quite insidious for two main reasons. First, the symptomatology (otorrhoea, otalgia, and hearing loss) can initially be almost identical to simple inflammatory or infective conditions of the ear, and often the main clue to the diagnosis can be its nonresolution. Second, spread to surrounding structures can occur very early and without much increase in symptoms, leading to the relatively advanced stage at which these tumors are often diagnosed.
The most common malignancy of the temporal bone by far is squamous cell carcinoma (SCC), and this is thus the focus of the discussions in this chapter. Other malignant pathologies arising from the temporal bone include adenocarcinoma, basal cell carcinoma, adenoid cystic carcinoma, sarcoma, melanoma, and metastases.1 Reported malignancies involving (but not arising from) the temporal bone include parotid tumors, bony tumors, soft tissue tumors, and tumors of the skin and skin appendages.
It is becoming increasingly clear that early aggressive surgical resection with adjuvant radiotherapy is important to maximize each patient’s outcome. However, such resections and treatment are major undertakings and need to be considered carefully in the clinical context.
Epidemiology
The incidence of SCC of the temporal bone has been reported to be approximately 1 per 1,000,000 per year in the United Kingdom for females and 0.8 for males.2 However, reports in the United States vary between 1 and 5 per 1,000,000 per year.3–5 Clearly the rarity of this tumor hinders accurate estimation. No predominance in racial groups has been identified.
There are differing ratios of gender distribution in different series. Several series reported a slight preponderance in males6–9; Morton et al2 and Gillespie et al5 reported a higher incidence in females; and in Moffat et al’s series,10 there was an equal distribution between genders. The most recent large series by Gidley et al11 comprised 53% males. Hence, it is likely that there is no real gender predilection.
Most series agree on a typical age of onset between 50 and 70 years of age.12 But it can occur in a younger age.
Etiology
Numerous studies have examined various etiological factors. While the complete picture remains unknown, two factors have clearly been reported as likely causes of temporal bone SCC:
1. Radiotherapy to nearby structures has been implicated in several series.13,14 The most commonly reported of these is radiotherapy for the treatment of nasopharyngeal carcinoma, however, intracranial radiotherapy has also been implicated. The latent period between radiotherapy exposure and the diagnosis of temporal bone carcinoma has been reported to be as long as up to 30 years, with a mean of between 12 and 15 years. Clearly this latency means that a thorough history of past treatment is essential with any suspected lesion.
2. Chronic inflammation and discharge, usually as a result of chronic suppurative otitis media (CSOM), is well established and often coexists with SCC of the temporal bone.6,15 This is considered to cause an irritation to the squamous epithelium lining the external auditory canal (EAC) and over time to be carcinogenic, although causation has not been definitively proved.
Other previously suggested etiological factors that have not gathered recent support include occupational radiation exposure (e.g., watch dial makers), exposure to chlorinated disinfectants,16 and ultraviolet exposure.
Clinical Features
The symptoms and signs of this disease are mostly correlated with the direct extent or spread of the tumor.
Symptoms
Otorrhoea, otalgia, hearing loss, and facial palsy account for the bulk of presenting symptoms. The frequency of each of these symptoms has been reported by several different series (Table 33.1).5–7,9–11,17–19 Bloody otorrhoea and severe pain should raise the index of suspicion and may help distinguish malignancy from simple CSOM.
Signs
Clearly the most definitive clinical finding is that of a raised, exophytic, irregular mass in the EAC or pinna (Fig. 33.1). However, this can also take the form of a small ulcer, polyp, or a relatively sessile lesion. Any such lesion warrants a timely biopsy to exclude carcinoma. These masses can be difficult to distinguish from severe CSOM purely clinically.
Subtle facial nerve palsy may not be reported by the patient, but this clinical finding is important to document as it is potentially a marker of the extent of disease and is a significant factor for staging (see below).
Table 33.1 Average Frequency of Clinical Symptoms at Presentation over Nine Series
Symptom | Average Frequency (%) |
Otorrhoea | 67 |
Otalgia | 58 |
Hearing loss | 43 |
Facial palsy | 23 |
Other cranial nerve palsies, while uncommon at presentation, should be actively looked for during examination.
Palpation of the neck is mandatory as metastasis to lymph nodes is a very poor prognostic sign.
Sites
Up to more than 90% of these tumors are located in either the EAC or on the pinna.20 This is made of approximately 60 to 80% arising on the pinna, with a further 12 to 30% from the external canal. Accurate statistics are hampered by the fact that quite often tumors cross these boundaries by the time of presentation. Only 1 to 10% arise from the middle ear cleft.21
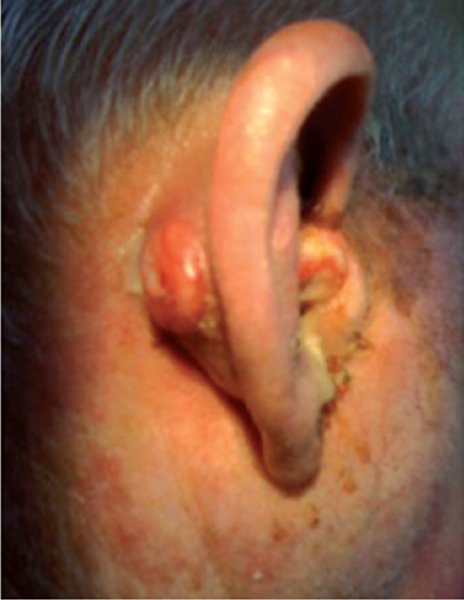
Figure 33.1 Large left temporal bone squamous cell carcinoma filling the external auditory canal and extensively ulcerating the pinna cartilage including the conchal bowl and the preauricular skin.
Pattern of Spread and Local Anatomy
Direct extension into local structures is the most common type of spread of temporal bone SCC.
Direct Extension
Extension in all directions is possible (Fig. 33.2). Because this region is densely packed with important anatomical structures, concomitant symptoms can be significant depending on the direction of extension (Table 33.2).
Further Spread
The lymphatic drainage network of the ear is not well developed and hence spread to lymph nodes is relatively uncommon at presentation, at approximately 10 to 23%.22 Hematological spread is even rarer and would make the disease incurable. Metastases to bone, lung, and liver can occur, but mortality usually ensues from locoregional disease progression.23
Perineural invasion is often a feature of SCC and the facial nerve’s convoluted course through the temporal bone makes it a prime target.24 This explains the relatively common presence of facial paresis or palsy on presentation.
Staging
The modified Pittsburg (2002) staging system24 (Table 33.3) is now the widely accepted classification scheme. This enables a useful direct comparison of outcomes from different centers from around the world. Moffat et al,10 Moody et al,18 Nyrop and Grøntved,17 Gidley et al,11 and others have used this system for reporting their series. Most have confirmed that its correlation with prognosis is better than previous systems.
The main modification from the original Pittsburg system (1990) was the addition of facial paresis at presentation as a feature indicating T4 disease. As lymph node involvement is a key poor prognostic indicator, any N1 disease immediately upstages the tumor to stage IV.
The first proposed staging system was by Stell and McCormick25 in 1985 based on the site of the origin of the tumor and the extent of local spread. This was soon modified by Clark et al26 in 1991 to add a T4 category for the involvement of dura or skull base, which had previously been included as T3 (Table 33.4). Around the same time, Arriaga et al27 developed the initial University of Pittsburgh Staging System (1990) to conform with the American Joint Committee on Cancer guidelines, which was modified in 2002 by Hirsch to its present state.
Diagnosis
After a detailed history and examination including careful cranial nerve assessment, the definitive diagnosis is made by direct biopsy of the lesion. If any doubt exists as to the nature of a mass encountered in the EAC, biopsy is strongly recommended instead of surveillance, as early diagnosis is paramount to a good prognostic outcome.
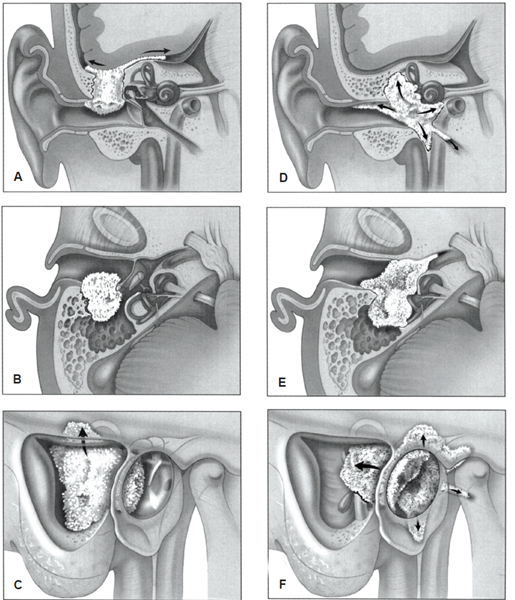
Printed with permission from: Lippincott William & Wilkins. Moffat DA, Chiossone-Kerdel JA, Da Cruz M. Squamous cell carcinoma. In: Jackler, Driscoll, eds. Tumors of the Ear and Temporal Bone. 2000:71; Fig. 18.3.
Table 33.2 Direction of Direct Extension of Temporal Bone SCC, Structures Affected, and Potential-Related Clinical Features
Direction of Spread | Structures Entered | Clinical Effects |
Lateral | EAC skin and vessels | Bloody otorrhoea EAC mass Otalgia |
Anterior | Temporomandibular joint Parotid Infratemporal fossa Petrosquamous fissure | Cheek swelling Trismus |
Superior | Epitympanum Tegmen tympani Middle fossa dura Temporal lobe | Temporal headache |
Posterior | Mastoid cells Posterior fossa dura | Postauricular mass Headache |
Inferior | Jugular foramen Vertebrae Cervical soft tissue | Cranial nerve palsies Neck swelling |
Medial | Tympanic membrane Middle ear cleft Facial nerve Eustachian tube Internal acoustic meatus Petrous apex Internal carotid artery | Facial palsy Conductive hearing loss Sensorineural hearing loss Vertigo and balance disturbance |
EAC, external auditory canal; SCC, squamous cell carcinoma.
Table 33.3 Modified Pittsburgh Staging System (2002)
Staging | Extent of Tumor |
T1 | Limited to the EAC without bony erosion or evidence of soft-tissue involvement |
T2 | Limited to the EAC with bony erosion (not full thickness) or limited soft-tissue involvement (< 0.5 cm) |
T3 | Erosion through the osseous EAC (full thickness) with limited soft-tissue involvement (< 0.5 cm), or tumor involving the middle ear, mastoid, or both |
T4 | Erosion through the cochlea, petrous apex, medial wall of the middle ear, carotid canal, or jugular foramen or dura; or with extensive soft-tissue involvement (> 0.5 cm), such as involvement of the TMJ or stylomastoid foramen; or with evidence of facial paresis |
N & M | As per AJCC TNM |
Stage I | T1N0 |
Stage II | T2N0 |
Stage III | T3N0 |
Stage IV | T4N0; or any T with N1–N3; or M1 |
AJCC, American Joint Committee on Cancer; EAC, external auditory canal; TMJ, temporomandibular joint; TNM, tumor-node-metastasis.
Table 33.4 Clark’s Modified Version of Stell’s Proposed Staging System (1991)
Staging | Extent of Tumor |
T1 | Tumor limited to the site of origin |
T2 | Tumor extending beyond the site of origin indicated by facial paralysis or radiological evidence of bone destruction |
T3 |



