24
Benign Neoplasms of the Temporal Bone
Lawrence R. Lustig and Robert K. Jackler
The benign tumors of the temporal bone comprise a diverse spectrum of lesions that have largely been responsible for defining the specialty of neurotology and skull base surgery. In spite of their benign histopathologic characteristics, however, these lesions may be locally destructive. Prompt diagnosis and treatment is therefore necessary to prevent further worsening of audiologic, vestibular, facial, or lower cranial nerve dysfunction that are so common upon presentation (Table 24–1).
 Schwannoma
Schwannoma
The most common tumor of the temporal bone and cerebellopontine angle is the schwannoma, accounting for 6% of all intracranial tumors, and 91% of all tumors in and around the temporal bone.1,2 Schwannomas are benign tumors of the nerve sheath, which historically have also been referred to as neuromas, neurofibromas, neurinomas, and neurilemmomas.3 Within the temporal bone, schwannomas arise in three anatomic loci: the internal auditory canal (IAC) from the eighth cranial nerve, the fallopian canal from the seventh cranial nerve, and the jugular foramen from cranial nerves IX to XI.
Vestibular Schwannoma/Acoustic Neuroma
Vestibular schwannomas, more commonly known as acoustic neuromas (ANs), are the most commonly occurring schwannoma of the temporal bone, and by inference, the most commonly encountered tumor in otology. An overwhelming majority of ANs arise de novo as a solitary lesion. Diagnosis typically occurs after the sixth decade, with a slightly higher incidence in females.4 Neurofibromatosis type 2 (NF2), accounting for only 5% of tumors, is associated with bilateral ANs and tends to present earlier in life (Fig. 24–1). Recent studies suggest that undiagnosed ANs may be present in as much as 2/10,000 of the population.5 Schwannomas, as their name implies, are derived from Schwann cells. Their point of origin is repeatedly described in the literature as arising at the transition zone between central and peripheral myelin, known as the Obersteiner-Redlich zone, though one small series indicates that most vestibular nerve schwannomas may in fact originate lateral to the glial-schwannian junction of the nerve.6,7 The tumors arise with an equal frequency from the superior and inferior divisions of the vestibular nerve, and usually originate within the medial portion of the IAC, though a fraction arise extrameatally or in the lateral IAC.8
The elucidation of the underlying genetics of ANs is derived from the study of NF2 patients. The specific defect for NF2 leading to bilateral ANs has been genetically mapped to chromosome 22.9 The gene product, termed merlin, is believed to be a tumor-suppressor gene, requiring both copies of the gene to be dysfunctional for tumorigenesis to occur. NF2 patients are therefore born with one defective gene, leading to a lifelong propensity toward AN development. Patients with sporadically arising ANs, by contrast, have acquired defects of both gene copies, leading to the formation of tumor.10,11 The precise role of the merlin gene product remains unclear, but has been shown to exert its activity by inhibiting phosphatidylinositol 3-kinase.12,13
| Site | Benign | Malignant | ||
|---|---|---|---|---|
| Pinna | Hemangioma | Basal cell carcinoma | ||
| Squamous cell carcinoma | ||||
| Melanoma | ||||
| EAC | Osteoma | Squamous cell carcinoma | ||
| Neurofibroma | Adenoidcystic carcinoma | |||
| Middle ear | Adenoma | Squamous cell carcinoma (rare) | ||
| Glomus tympanicum | Rhabdomyosarcoma | |||
| Schwannoma (CN VII) | ||||
| Mastoid | Adenoma | Squamous cell carcinoma (rare) | ||
| Schwannoma (CN VII) | Papillary adenocarcinoma | |||
| IAC | Schwannoma (CN VIII>>VII) | |||
| Meningioma Ossifying hemangioma | ||||
| Jugular foramen | Glomus jugulare Meningioma | |||
| Schwannoma (CN IX-XII) | ||||
| Petrous apex | Chondroma | Chondrosarcoma | ||
| Chordoma | ||||
| Metastases | ||||
EAC, external auditory canal; IAC, internal auditory canal.
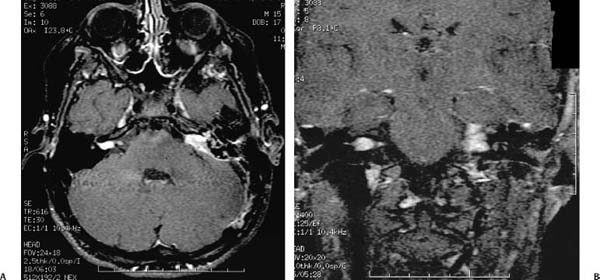
Figure 24–1 A patient with neurofibromatosis type 2 (NF2) and bilateral vestibular schwannomas. The T1-weighted MRI scan with gadolinium enhancement in the axial plane (A) shows a characteristic tumor of the right internal auditory canal with a small cerebellopontine angle component. The tumor on the left, which has the appearance of a meningioma with dural tail enhancement, was found to be a vestibular schwannoma at surgery. The coronal view (B) shows the tumors in this same patient.
Macroscopically, ANs are smooth-walled gray or yellowish masses. Though they have been traditionally described as being well encapsulated, studies indicate that they do not possess a true capsule.14 Microscopically, two morphologic patterns can be discerned. The Antoni A pattern consists of densely packed spindle-shaped cells with darkly staining nuclei. When they appear in a whorled configuration, it is referred to as a Verocay body. The Antoni B pattern consists of a more diffusely arranged cell pattern with increased pleomorphism (Fig. 24–2). Any tumor may contain one or both patterns. The clinical significance of these two patterns is unclear, though the Antoni B type tends to predominate in larger tumors. The immunoperoxidase stain S-100 is positive and is used to confirm the diagnosis of schwannoma.15–17
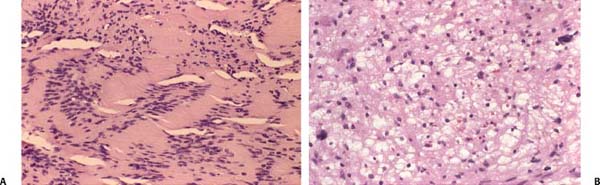
Figure 24–2 Histopathology of vestibular schwannomas. These hematoxylin and eosin (H&E) stains demonstrate the two common types of histopathology seen in vestibular schwannomas. The Antoni A pattern (A) consists of densely packed spindle-shaped cells with darkly staining nuclei. When they appear in a whorled configuration it is referred to as a Verocay body. The Antoni B pattern (B) consists of a more diffusely arranged cell pattern with increased pleomorphism. This pattern tends to predominate in larger tumors, though any tumor may contain one or both patterns. The clinical significance of these two patterns is unclear.
The majority of ANs are benign and slow-growing tumors. Though the average growth rate for tumors has been estimated to be between 0.1 and 0.2 cm in diameter per year, the range is variable and 10 to 15% have a growth rate greater than 1 cm per year.18,19 The growth rate has been shown to be related to the concentration of vascular endothelial growth factor.20 The tumors usually originate in the IAC. Growth then carries the tumor into the cerebellopontine angle cistern, where it commonly involves the seventh and eighth cranial nerves. Further enlargement causes brainstem compression and fifth nerve involvement, and eventually hydrocephalus.
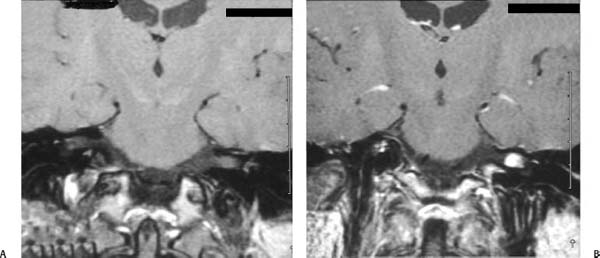
Figure 24–3 A coronal MRI of a small left intracanalicular vestibular schwannoma measuring about 8 mm. The tumor is barely visible on the T1-weighted images without gadolinium enhancement (A), whereas with gadolinium (B) the tumor is markedly enhanced.
The clinical presentation of patients with AN reflects the tumor growth pattern. Asymmetrical sensorineural hearing loss, occurring in 95% of patients, is believed to be secondary to direct compression of the tumor on cranial nerve VIII within the IAC, or due to compression of the nerve’s vascular supply. The hearing loss is sudden in onset in about one fourth of cases.4 Additional symptoms include high-pitched, continuous, asymmetrical tinnitus; vertigo; disequilibrium and ataxia (up to 70% incidence in larger tumors); facial sensory disturbances (50%); facial twitching (10%); headaches (40%); nystagmus; and decreased corneal reflexes.4 Audiometric testing typically reveals asymmetrical sensorineural hearing loss predominating in the high frequencies, though this configuration is not strictly found. Speech discrimination scores often are out of proportion to the degree of pure-tone hearing thresholds. There is usually either an absent stapedial reflex or reflex decay, but this is not sufficiently reliable to be of much diagnostic value.21 Auditory brainstem response (ABR) testing is also used to assist in identifying retrocochlear pathology. The presence of a wave I and absence of waves II to V is the most specific finding for AN, though one must be wary for both false-positive (>80%) and false-negative (12–18%) ABRs.22,23
Contrast-enhanced magnetic resonance imaging (MRI) provides the gold standard for the diagnosis of the AN, which is able to detect tumors as small as 1 mm. The well-demarcated lesions are isointense on T1-weighted images and demonstrate some signal increase on T2-weighted images with areas of heterogeneity24 (Fig. 24–3). After gadolinium administration, enhancement is striking, more so than most other benign extraaxial tumors.25 High-resolution computed tomography (CT), though not as sensitive as MRI for small tumors, reliably demonstrates a smoothly marginated, contrast-enhancing mass within the cerebellopontine angle (CPA) in tumors over 1.5 cm in diameter25 (Fig. 24–4).
Although the tumors are slow growing and benign, in mostcases treatment is recommended because growth may lead to multiple cranial neuropathies, brainstem compression, hydrocephalus, and death. In selected cases, a conservative “watch and wait” approach may be appropriate, such as in the elderly or medically infirm.26,27 The first priority of surgery or radiation therapy is to alleviate the risk of progressive intracranial tumor growth, and it is secondarily concerned with preservation of facial nerve function and sparing of useful hearing. A variety of techniques have been employed to achieve these ends, including the translabyrinthine, retrosigmoid, and middle fossa approaches. The decision of which to use depends on the tumor size, its depth of penetration within the IAC, the degree of hearing loss, and the experience of the surgical team (Table 24–2).28,29 A recent surgical trend also includes near-total tumor resection (remnant ≤2.5 mm in length and ≤2 mm thick) followed by expectant observation in an effort to improve facial nerve outcomes.30 In such a scenario, recurrences have been shown to be about 3%. Intraoperative cranial nerve monitoring is routinely employed to assist with neural preservation during tumor resection. The results after surgery are dependent on the experience of the surgical team.31 The expected mortality is less than 2% in most major centers, with tumor-related mortality limited to those with large tumors. Complications occur in about 20% of cases, and most commonly include cerebrospinal fluid (CSF) leakage, meningitis, and chronic headache.32–34 Less common are traumatic parenchymal injury from intraoperative retraction, arterial or venous cerebral infarct, postoperative hemorrhage into the CPA, and air embolism.35,36 Anatomically the facial nerve is preserved in 82 to 97% of cases, with an overwhelming majority having grade 1 or 2 facial nerve function 1 year after surgery. Whether anatomic preservation correlates with postoperative nerve function, however, is subject to debate.37,38 Hearing preservation surgery may be attempted for tumors with less than a 1.5 cm intracranial component, and that meet the “50/50” rule, speech reception threshold less than 50 dB and a speech discrimination score of greater than 50%, though these rules are not strict and are even now being redefined and broadened.39–42 Though results vary widely from center to center, useful hearing is commonly preserved in about one fourth of cases attempted, though in the most favorable tumors hearing preservation may be as high as 70% in experienced centers.43,44
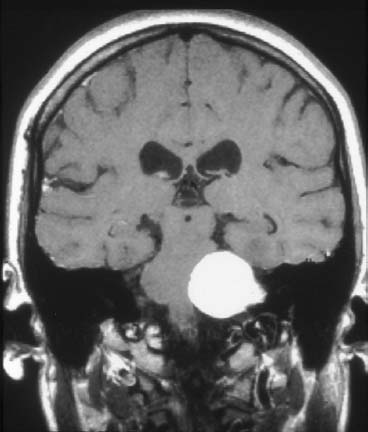
Figure 24–4 A 4-cm vestibular schwannoma with brainstem compression is shown in this coronal T1-weighted MRI scan with gadolinium enhancement.
Stereotactic radiosurgery (“gamma-knife”) is being increasingly employed as an alternative to surgery in a growing number of centers with acceptable morbidity and a similar spectrum of functional deficits, though the long-term control rates have not yet been conclusively established. For those unable to tolerate the risk of surgery, radiation may represent a viable alternative.45–49 There is also growing acceptance that for large tumors, a subtotal resection of tumor, leaving the tumor capsule behind to preserve existing cranial nerve function, followed by radiation or gamma knife treatment, represents an alternative treatment option.50 Several larger series with adequate (10 years or greater) follow-up have shown that following single dose radiotherapy, 20 to 25% of tumors remain stable, 50 to 75% of tumors shrink in size, and 2 to 13% show further growth.51–54 However, these rates need to be tempered by the observation that in untreated tumors followed over a 3-year period, 50% of tumors remain stable, 14% shrink, and 37% enlarge.55 Though older dosing regimens were associated with a 37% incidence of facial palsy, newer dosing algorithms are rarely associated with facial nerve weakness following stereotactic radiotherapy.53 Preservation of useful hearing (>50% speech reception threshold and 50% word recognition scores) has been shown to occur in 47 to 79% of radiation tumors.53,54,56,57 However, should a patient undergo stereotactic radiotherapy for a vestibular schwannoma and subsequently need surgical resection for a continually growing tumor, the facial nerve function preservation rates are not as good as compared with when there was no prior radiotherapy.58–60 Further, because of the risk of a radiation-induced malignancy, radiotherapy for these lesions should only be cautiously used in younger individuals.61
| Approach | Advantages | Disadvantages | ||
|---|---|---|---|---|
| Retrosigmoid | Excellent exposure | Increased incidence of postop headaches | ||
| Hearing preservation possible | Higher incidence of CSF leak | |||
| Need for more vigorous cerebellar retraction | ||||
| Translabyrinthine/anterosigmoid | Lower surgical morbidity | Inability to preserve hearing | ||
| More facial nerve reconstructive options | ||||
| Middle fossa | Superior hearing preservation results | Increased risk of transient facial neuropraxia | ||
| Unsuitable for tumors with large CPA component | ||||
CPA, cerebellopontine angle; CSF, cerebrospinal fluid.
The major dilemma in vestibular schwannoma management is a young healthy patient with good hearing and an intracanalicular tumor: to treat or wait and rescan in 6-12 months. First, the patient must understand that the least risk to hearing is to do nothing; however, even small tumors can suddenly cause hearing to drop. Also, in theory, facial nerve injury during surgery is greater if the tumor grows in the 6-12 month follow-up interval. Even the risk of partial but significant hearing loss with gamma knife (about 25% chance) outweighs most other considerations. Wait and re-scan has become the standard for initial management of intracanalicular schwannomas for many neurotologists. One major exception is significant dizzy attacks, which are best managed by tumor surgery which sections the vestibular nerve.
Jugular Foramen Schwannoma
Though schwannomas are the second most common lesion of the jugular foramen behind glomus tumors, overall they are relatively rare, representing about 3% of all intracranial schwannomas.62,63 In fact, the largest series, that of Tan 62 in 1990, includes only 14 patients, with less than 100 cases in the world literature reported up to that time. Schwannomas presenting in this region arise from cranial nerves IX to XII. As with vestibular schwannomas, these tumors probably occur at the transition zone between the central and peripheral myelin. Histologically, the tumors resemble vestibular schwannomas.64
Three tumor growth patterns have been recognized for jugular foramen schwannomas.65 Tumors arising in the distal portion of the foramen may expand inferiorly out of the skull base. More proximally arising tumors can expand into the posterior fossa. Others arise in the middle of the foramen and either expand primarily into bone or become bilobed, with an expansion both out of the skull base and into the posterior fossa.
The most common presenting symptoms are hoarseness, swallowing difficulties, and vertigo.62–65 Other symptoms may include shoulder weakness, headache, nausea, vomiting, facial numbness or spasm, dysphagia, and visual disturbances. On exam, cranial nerve X is dysfunctional in 63% of cases presenting, and may be accompanied by deficits of cranial nerves IX (55%), XI (41%), and XII (36%).66 Cranial nerve V and VII dysfunction is less common on examination, as are hemifacial spasm, nystagmus, ataxia, and papilledema.62
High-resolution CT typically demonstrates a well-demarcated, smoothly marginated expansion of the foramen walls (Fig. 24–5). MRI is superior for diagnosis, and demonstrates a lesion isointense to brain parenchyma on contrast-enhanced T1-weighted images, whereas T2-images usually reveal a high signal intensity. The addition of gadolinium causes a marked signal increase. Differentiation from paragangliomas is made possible by noting the morphologically smooth manner of bony erosion as compared with a more irregular pattern with glomus tumors and meningiomas. In contrast to glomus tumors, flow voids are notably absent.63 Angiography is often undertaken under the presumption that the tumor is a paraganglioma, and has little use diagnostically for schwannomas unless one anticipates a possible surgical need to evaluate the carotid or jugulosigmoid venous systems.63
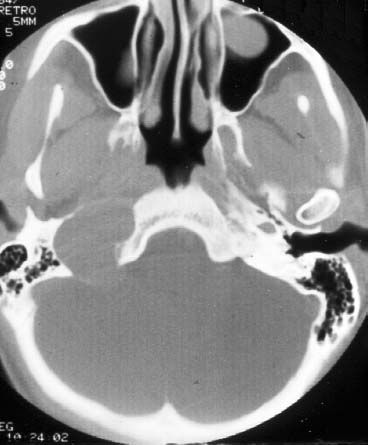
Figure 24–5 An axial CT scan of a right-sided jugular foramen schwannoma. Note the smooth enlargement of the jugular foramen. The patient presented with headaches, hoarseness, and dysphagia.
As with vestibular schwannomas, the treatment is primarily surgical.67 Because of the variability of tumor presentation, the surgical approach must be individualized. Techniques for exposure of the jugular foramen have become increasingly routine in recent years.68,69 Jugular foramen schwannomas, because they often possess an intracranial component, frequently require a transjugular posterior fossa craniotomy. Complete removal often causes paralysis of cranial nerves IX to XI, leading to postoperative hoarseness, dysphagia, and shoulder weakness in many cases.66 Vocal cord medialization procedures can help compensate for paralytic laryngeal dysfunction.
Facial Nerve Schwannoma
Schwannomas of the facial nerve are uncommon lesions, accounting for only 1.2% of all temporal bone tumors.1 Though its true incidence is not known, one study was able to identify only one case out of 1400 temporal bones analyzed.70 Schwannomas have been identified along the entire course of the facial nerve, although intratemporal tumors appear to be much more common than the intracranial variety.71,72 Within the temporal bone, the most common sites of involvement, in decreasing frequency, are geniculate ganglion, horizontal and vertical segments, IAC, and labyrinthine segment. A small percentage, however, display an unusual multicentricity evidenced by multiple discrete intraneural connections, sometimes described as a string of pearls.73 The tendency for growth longitudinally along the lumen of the fallopian canal may lead to tumor prolapse into the middle ear, IAC, and CPA, and out of the stylomastoid foramen.
In contrast to ANs, facial nerve schwannomas tend to be slower growing and are often present for years before detection.74 However, because of the facial nerve’s intimate relationship with the sensory organs, otic capsule erosion is more common, occurring in up to 30% of cases.72
Facial nerve dysfunction (palsy or twitch) is the hallmark of the clinical presentation. It occurs due to compression of the nerve within the fallopian canal. The most common pattern is slowly progressive palsy, often accompanied by hyperfunction manifested as limited twitch or full hemifacial spasm. Recurrent acute paralytic episodes with partial or even complete recovery may also occur. Patients are commonly misdiagnosed with Bell’s palsy with the first episode of paralysis. Successive bouts of palsy then ensue, with increasingly poorer facial nerve function. This presentation of recurrent, progressively more severe episodes of facial palsy is a classic characteristic of facial nerve schwannoma. The facial nerve is surprisingly resistant to compression. It has been estimated that 50% of facial nerve fibers must degenerate before clinical signs of a palsy are detected.74,75 In one study of 48 patients with facial nerve neuromas, 26 presented with normal facial function.72 Thus, patients without functional recovery from an idiopathic facial paralysis after 3 months or with a history of recurrent Bell’s Palsy should have an enhanced MRI scan to search for tumor or facial nerve pathology.74 Patients may also present with normal facial nerve function and a conductive hearing loss.72 Additional presenting symptoms include vertigo from a labyrinthine fistula and sensorineural hearing loss from cochlear invasion.56,76–78 Prolonged pain should also raise one’s suspicion for a diagnosis other than idiopathic facial palsy.72–74 Examination of the ear may demonstrate a mass behind the drum in up to 29% of cases.74 Because biopsy of a facial nerve schwannoma in the middle ear usually results in a facial paralysis, appropriate imaging studies are recommended prior to biopsy of any middle ear tumor. Site of lesion tests, such as the Schirmer’s test of lacrimation and stapedial reflex testing, while theoretically attractive, are not completely reliable and have been made largely obsolete by CT and MRI.
Radiographically, facial nerve schwannomas are similar to those arising in other portions of the temporal bone. They are hypointense on T1 images, hyperintense on T2 images, and show marked enhancement with gadolinium. An enhancing enlargement of varying thickness along a large segment of facial nerve is considered highly suggestive of schwannoma. Although high-resolution CT can identify these tumors due to their osseous erosion, MRI is a more sensitive diagnostic tool.79
The treatment for facial nerve schwannomas is primarily surgical.51,76–78 The primary goal in management of an intratemporal facial nerve schwannoma is maintenance of facial function. With good facial function, it is usually best to leave the tumors alone as resection and grafting lead to at best a House-Brackmann grade of 3/6 (facial weakness at rest with good eye closure). For lesions limited to the transverse or descending portions of the nerve, a tympanomastoid approach may be used.80 Lesions that involve the labyrinthine segment, IAC, or geniculate ganglion require the addition of an extradural middle cranial fossa approach. If cochlear function has been destroyed, then a translabyrinthine approach may be utilized.74 At surgery, it is occasionally possible to remove a facial nerve schwannoma with preservation of its nerve of origin. More commonly, however, nerve repair with an interposition graft is needed. This may be accomplished with either a greater auricular or sural nerve graft. In general, those patients with long-standing facial nerve paralysis (>12 months) tend to have poorer postoperative facial nerve function. Because a common presentation for facial nerve schwannomas is a conductive hearing loss, it is not uncommon to first identify these tumors intraoperatively during an exploratory tympanotomy with the intent to perform a stapedectomy. In such a scenario, if a soft tissue mass is identified leaning on or eroding the stapes superstructure at tympanotomy, the surgeon should halt the procedure and perform imaging studies, and not biopsy the lesion.
 Paraganglioma (Glomus Tumor)
Paraganglioma (Glomus Tumor)
The most common tumor of the middle ear and second most common tumor found in the temporal bone is the paraganglioma, more commonly known as a glomus tumor but occasionally referred to as a chemodectoma.81 Paraganglia, the origin of these tumors, exist throughout the temporal bone, including on the jugular dome, the promontory of the middle ear, and along Jacobson’s and Arnold’s nerves, and account for the predilection of glomus tumors toward these anatomic sites.82 The term glomus was mistakenly attached to these tumors when it was believed that their origin was similar to true glomus (arteriovenous) complexes, and though now recognized as inaccurate, the nomenclature has persisted.83
Although most glomus tumors appear to arise sporadically, there are reports of families with several members affected by glomus tumors, with an unusual genomic imprinting mode of inheritance.84,85 In this manner of transmission, tumors only occur in the offspring of an affected female when there is transmittance of the gene through a carrier male, accounting for the observed tumor occurrence in “skipped” generations.86 A genetic marker for familial paragangliomas has been localized to chromosome 11, though a precise genetic cause has not been identified as of yet.85 There is a clear predilection for these tumors to arise in females, and patients usually present after the fifth decade of life.81–84,86,87
Glomus tumors are typically reddish-purple, vascular, and lobulated masses. Histologically they resemble normal paraganglia with clusters of chief cells, characteristically termed zellballen (literally translated as “cell balls”) in a highly vascular stroma. This pattern is enhanced on silver staining, which is useful diagnostically. Sustentacular cells and nerve axons, seen in the normal paraganglion, are rarely seen in the tumor, however.88,89
Glomus tumors contain the neural crest cell-derived chief cells, which are included in the diffuse neuroendocrine system (DNES). As a result, they have the potential to produce catecholamines, producing a physiologic response similar to pheochromocytomas. Fortunately, this is extremely rare, occurring in only 1 to 3% of glomus tumors.83,89,90 Nevertheless, it is reasonable to perform preoperative evaluation for the presence of catecholamine-producing tumors in all patients, because life-threatening intraoperative hypertension is possible. Elevation of urine catecholamine levels (three to five times normal) requires differentiation from pheochromocytomas, and occasionally may require selective renal vein sampling for adequate diagnosis.91
Glomus tumors involving the temporal bone are divided into two categories based on their anatomic location. Other classification schemes further subdivide these tumors according to size and extent of invasion (Table 24–3).91 Those arising along the course of Jacobson’s nerve and involving primarily the tympanic cavity are termed glomus tympanicum. Paragangliomas arising from the dome of the jugular bulb and involving the jugular foramen and related structures are termed glomus jugulare. Both types are marked by slow, progressive growth, spreading via the pathways of least resistance, such as the temporal bone air-cell tracts, neural foramina, vascular channels, bony haversian systems, and the eustachian tube.91–94 Advanced lesions of either type have the ability to invade cranial nerves.95 However, the clinical presentation and operative management of each may be markedly different, and thus each is discussed individually. Glomus vagale tumors arise beneath the cranial base in proximity to cranial nerve X. A small minority of vagale tumors involve the temporal bone via retrograde spread through the jugular foramen.
The appearance of a paraganglioma on MRI reflects its highly vascular nature. Glomus tumors are isointense on T1-weighted images and brightly enhance with gadolinium. They typically possess numerous signal voids due to the numerous vascular channels within them. On T2-weighted images, they demonstrate increased signal intensity in the solid portions of the tumor with persistent flow void in the vascular portions.24 Because paragangliomas can be multiple, some advocate that the imaging study should be carried down to the level of the carotid bifurcation to determine if multiple tumors exist.96 Angiography is an additional important aspect of the evaluation of glomus tumors, but should be deferred until the preoperative period when both diagnostic and therapeutic (embolization) measures can be accomplished in a single study. The study allows the determination of arterial supply, degree of vascularity, degree of arteriovenous shunting, evidence of major venous sinus occlusion, and confirmation of the diagnosis another advantage of angiography is that it can single-handedly evaluate both the internal and external carotid systems for evidence of multiple early lesions. Embolization is usually performed at the time of angiography as a preoperative maneuver to limit surgical blood loss.97,98 Magnetic resonance angiography and venography are newer modalities that can also aid in the diagnosis of vascular lesions of the temporal bone including glomus tumors. The role of these newer radiographic modalities in the evaluation of glomus tumors is currently being defined.99
| Glasscock/Jackson Classification of Glomus Tumors | ||
|---|---|---|
| Tumor | Description | |
| Glomus tympanicum | ||
| Type I | Small mass limited to the promontory | |
| Type II | Tumor completely filling the middle ear space | |
| Type III | Tumor filling the middle ear and extending into the mastoid | |
| Type IV | Tumor filling the middle ear, extending into the mastoid or through the tympanic membrane to fill the external auditory canal; +/—internal carotid artery involvement | |
| Glomus jugulare | ||
| Type I | Small tumors involving the jugular bulb, middle ear, and mastoid | |
| Type II | Tumor extending under the internal auditory canal; might have intracranial extension | |
| Type III | Tumor extending into petrous apex; might have intracranial extension | |
| Type IV | Tumor extending beyond petrous apex into clivus or infratemporal fossa; might have intracranial extension | |
| Fisch Classification of Glomus Tumors | ||
| Type A | Tumors limited to the middle ear cleft (Glomus tympanicum) | |
| Type B | Tumors limited to the tympanomastoid area with no bone destruction in the infralabyrinthine compartment of the temporal bone | |
| Type C | Tumors involving the infralabyrinthine compartment with extension into the petrous apex | |
| Type D1 | Tumors with intracranial extension ≤2 cm in diameter | |
| Type D2 | Tumors with intracranial extension >2 cm in diameter | |
Sources: Jackson CG. Skull base surgery. Am J Otol 1981;3:161–171; Oldring D, Fisch U. Glomus tumors of the temporal region: Surgical therapy. Am J Otol 1979;1:7–18. Reprinted by permission.
Glomus Tympanicum
Glomus tympanicum is a paraganglioma that arises from the promontory of the middle ear. Because of the vascularity of these tumors, pulsatile tinnitus is often the first presenting symptom.92 Further growth causes conductive hearing loss as ossicular mobility is inhibited, which occurs in approximately half of all patients.91 Continued expansion may cause the glomus tympanicum to erode laterally through the drum, mimicking a friable, bleeding polyp, or it may expand medially causing facial nerve dysfunction, sensorineural hearing loss, or vertigo.81–84,86,88–93 Rarely, it may present as a eustachian tube mass or epistaxis.100,101 In one large series of 71 patients, presenting symptoms, in order of decreasing frequency, were pulsatile tinnitus (76%), hearing loss (conductive 52%, mixed 17%, sensorineural 5%), aural pressure/fullness (18%), vertigo/dizziness (9%), external canal bleeding (7%), and headache (4%).93 Brown’s sign, which consists of a pulsatile, purple-red middle ear mass that blanches with positive pneumatic otoscopy, is a frequently mentioned distinguishing sign but is of little clinical value.102
The differentiation between tympanicum and jugulare tumors is not always possible by physical examination alone because both lesions typically involve the middle ear.91 Furthermore, other vascular lesions of the middle ear, such as an aberrant carotid artery or a high-riding jugular bulb, may mimic a glomus tumor, and thus radiographic evaluation prior to biopsy or surgical intervention is important. Temporal bone CT can identify an intact plate of bone at the lateral aspect of the jugular fossa, indicating that a tumor is limited to the middle ear and aiding its identification as a glomus tympanicum. CT is also useful for evaluating the degree of bony erosion and the tumor’s relationship to surrounding temporal bone structures.91–93,95,96 MRI, although not as good evaluating bony changes within the temporal bone as CT, is superior in identifying the extent of the tumor and defining the relationship of tumor to surrounding structures once it has extended beyond the confines of the middle ear.103 Angiography, although useful for larger lesions, is not required for small glomus tympanicum tumors limited to the middle ear.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


