Rate of Growth
Generally, benign tumors grow slowly, whereas malignant tumors grow rapidly. The rate of growth of malignant tumors is inversely proportional to their degree of differentiation.
Local Invasion
Most benign tumors are localized, whereas malignant tumors have the potential to invade adjacent tissue or to metastasize to distant sites. Many benign tumors such as schwannoma have a true capsule that limits their growth (Fig. 14.1B). Others such as pleomorphic adenoma have a pseudocapsule composed of condensed connective tissue that gradually thickens with the slow expansion of the tumor (Fig. 14.1C). Some benign tumors possess neither of these. Malignant tumors do not possess capsules and infiltrate adjoining tissue (Fig. 14.1D), which necessitates excision of some normal surrounding tissue during surgery to ensure complete tumor removal.
Metastasis
The presence of metastasis alone is sufficient to label a tumor as malignant. Metastasis can be defined as the presence of tumor implants that are discontinuous from the primary tumor and located at a site that is distant from the tissue of origin. Malignant ocular and orbital tumors may metastasize by hematogenous spread (e.g., alveolar soft part sarcoma), lymphatic spread (e.g., squamous cell carcinoma, SCC), or perineural spread (e.g., adenoid cystic carcinoma). The pattern of lymph node involvement depends on the natural lymphatic drainage of the host tissue. For example, sebaceous carcinoma of the eyelid first metastasizes to the preauricular nodes. Skip metastases can sometimes be seen when the tumor cells lodge in the distal nodes without involving proximal nodes.
Controversy: A sentinel lymph node is the first regional lymph node to receive lymph flow from the primary tumor. Pathologic examination of such a node may help assess the extent of disease and plan further management and has been advocated by some for tumors of the eyelid and conjunctiva.28 Skip metastases may be missed with the sentinel node technique.
Carcinogenesis
Cancer is a multistep process during which cells undergo profound metabolic and behavioral changes that lead to excessive, untimely proliferation, escape from surveillance by the immune system, and ultimately invade distant tissues to develop metastases.29 Genetic damage is the cornerstone of carcinogenesis and may be caused by environmental factors, chemicals, viruses, or hormones.
Physical Carcinogenesis
Ultraviolet (UV) light and ionizing radiation are the most commonly implicated physical agents in carcinogenesis.27 In 1992, the International Agency for Research on Cancer (IARC) classified UV radiation as a Category I carcinogen, stating that solar radiation causes cutaneous malignant melanoma and nonmelanocytic skin cancer.30 UV light induces mutation, inactivation of enzymes, and sometimes cell death. At the biochemical level, exposure to UV light results in formation of pyrimidine dimers in the DNA that are usually repaired in normal individuals. In genetically susceptible individuals (e.g., those with xeroderma pigmentosa, XP), such damage remains unrepaired and predisposes to cancers on prolonged exposure to UV light. Ionizing radiation damages DNA by altering the cellular DNA or by formation of free radicals (Fig. 14.2). The DNA damage, in turn, leads to mutagenesis. Nonradiation physical carcinogenesis is rare and may result from longstanding stones or implants.

Chemical Carcinogenesis
Since the first report by Sir Percival Pott in 1775 on the higher incidence of scrotal cancer among chimney sweepers, several chemical carcinogens have been identified.31 Cellular transformation by chemical carcinogens involves three stages: initiation, promotion, and progression. Directly acting chemical carcinogens such as alkylating and acylating agents cause cellular transformation without a need for metabolic conversion, whereas indirectly acting carcinogens (procarcinogens) such as aromatic amines or azo dyes must be metabolically converted in the body to become carcinogenic. The directly acting and indirectly acting carcinogens contain electrophile groups that form adducts with DNA, RNA, and proteins. Although any gene can be affected, the RAS and TP53 genes are the most frequent targets of chemical carcinogens. After one cycle of proliferation, the DNA damage is transferred to the progeny cells, thus making it permanent and irreversible and susceptible to the action of promoters. Promoters such as hormones, drugs, and phenols are by themselves nontumorigenic but augment the carcinogenicity of chemicals. They also cause clonal proliferation and expansion of “initiated” cells, which now have a reduced requirement for growth factors. The “initiated” clones of cells accumulate additional mutations and proliferate to form a malignant tumor.
Viral and Microbial Carcinogenesis
Several viruses and microbial agents are known to cause cancers and are listed in Table 14.1. Human papilloma virus (HPV) causes squamous papillomas (types 1, 2, 4, 6, and 7) and cervical or anogenital SCC (types 16 and 18). Recent literature describes the role of HPV in ocular surface squamous neoplasia (OSSN).32 HPV owes its oncogenic potential to viral genes E6 and E7, which interact with growth-regulating proteins (Fig. 14.3).33 The E7 protein binds to the retinoblastoma protein and releases E2F transcription factors, which normally are sequestered by Rb, promoting progression through the cell cycle. The E6 protein binds to and causes degradation of p53. E7 also inactivates the cyclin-dependent kinases (CDKs) 1A/p21 and 1B/p27. In addition, the proteins E1 and E2 play an important role in viral genome replication.33 High-risk HPV types stimulate the loss of tumor suppressor genes, activate cyclins, inhibit apoptosis, and combat cellular senescence.
Table 14.1
Viruses and Human Cancers
| Virus | Associated Cancer | Mechanism of Action |
| Epstein-Barr virus (EBV) | Hodgkin lymphoma Burkitt lymphoma Nasopharyngeal carcinoma Non-Hodgkin lymphoma in immunocompromised states | NF-κB and JAK/STAT signaling pathways activation by LMP1 |
| Kaposi sarcoma virus | Kaposi sarcoma Primary effusion lymphoma | Production of viral oncoproteins |
| Human papilloma virus (HPV) | Cancers of the cervix, anal canal, vulva, vagina, penis | Production of E6 and E7 viral oncoproteins |
| Human T-cell leukemia virus – type 1 (HTLV1) | Adult T-cell leukemia/lymphoma | Viral oncoprotein TAX |
| Hepatitis B virus (HBV) | Hepatocellular carcinoma | Chronic irritation and inflammation |
| Hepatitis C virus (HCV) | Hepatocellular carcinoma | Chronic irritation and inflammation |
| Human immunodeficiency virus (HIV) | Kaposi sarcoma | Immunosuppression |

Epstein-Barr virus (EBV) was the first virus to be associated with human cancer and has now been associated with Burkitt lymphoma (BL), orbital T-cell lymphoma, and several other cancers.34 Latent membrane protein 1 (LMP1), an EBV-encoded gene, acts as an oncogene and promotes B-cell proliferation by activating nuclear factor kappa-B (NF-κB) and JAK/STAT signaling pathways. LMP1 also inactivates apoptosis by activating the B-cell lymphoma 2 (BCL2) gene.
Human T-cell lymphotropic virus-1 (HTLV-1) is the only retrovirus to be implicated in human cancers. HTLV-1, implicated in human T-cell lymphoma and leukemia, contains a pX region in its genome having the TAX protein necessary for cellular transformation. The TAX protein transactivates the expression of genes that encode cytokines, cytokine receptors, and co-stimulatory molecules, which sets up an autocrine system leading to T-cell proliferation. Further, TAX also represses tumor suppressor genes such as CDKN2A/p16 and TP53.
More recently, Merkel cell carcinoma polyomavirus has gained attention due to its association with Merkel cell carcinoma, a rare and aggressive cutaneous malignancy exhibiting neuroendocrine differentiation.35 Although research so far points toward a role of T antigen, the exact mechanisms of oncogenesis are not well defined.35
Helicobacter pylori was the first bacterium to be classified as a carcinogen. In addition to being a causative agent for peptic ulcer and gastric cancer, it is now associated with ocular adnexal lymphoma (OAL).36 Similarly, Chlamydia psittaci has been implicated in ocular adnexal mucosa-associated lymphoid tissue (MALT) lymphoma, possibly by clonal selection on MALT and subsequent lymphoma development.37 The exact mechanisms of oncogenesis are incompletely understood but possibly involve inflammatory cytokines that ultimately trigger clonal B-cell proliferation. The role of C. psittaci in OAL is still being debated. Although few authors disagree on this association, others have succeeded in demonstrating presence of C. psittaci in tissues of these patients.38,39 This belief has been further supplemented by regression of these lymphomas following antibiotic usage.40
Hormonal Oncogenesis
Several cancers in the human body (e.g., breast, testis, prostate, thyroid, and endometrium) are hormone related. In these cancers, the hormones act to induce cell proliferation and thus provide an opportunity of accumulation of random genetic errors.41 Recent literature has described expression of hormonal receptors in cancers of the conjunctiva and eyelids, thus suggesting a possible role of hormonal oncogenesis.42,43
Genetic Basis of Cancer
Genetic damage is the cornerstone of carcinogenesis. Clonal proliferation of a single progenitor cell inhabiting genetic damage results in the formation of a tumor. These cellular transformations may result from several alterations that affect cancer-associated genes.
Karyotypic Changes
Karyotypic changes in cancers can be subtle and remain undetected or may be large enough to be seen by light microscopy. Balanced translocations are seen in some hematopoietic and mesenchymal neoplasms. In BL, translocation between chromosome 8 and 14 result in over expression of the MYC gene on chromosome 8 as a result of juxtaposition with the immunoglobulin heavy chain gene on chromosome 14 (Fig. 14.4).44 Because the expression of several genes in the human body is regulated by c-myc, the constitutive expression results in cell growth, uncontrolled proliferation, and decreased apoptotic threshold. In follicular lymphoma (FL), translocation between chromosomes 14 and 18 lead to overexpression of the Bcl2 (antiapoptotic) gene.45 This constitutive expression of Bcl2 in the germinal center of B cells leads to accumulation of damaged B cells that have a prolonged lifespan and are ready for additional genetic damage, which, in turn, eventually leads to development of FL. The t(11;22)(q24;q12) translocation seen in Ewing sarcoma results in fusion of the EWS gene on chromosome 11 with the FLI1 gene on chromosome 22 to form EWS-FLI1, which promotes cell proliferation and survival.46

Deletion, the second most frequent karyotypic abnormality, may result in loss of tumor suppressor genes. A common mechanism for this is an inactivating point mutation in one allele, followed by deletion of the other, nonmutated allele.47 Such deletions result in loss of heterozygosity. Loss of heterozygosity involving 1p36 and 11q23 is seen in the majority of neuroblastomas and is associated with poor clinical outcomes.48
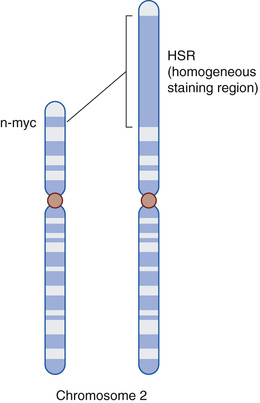
Gene amplification, which refers to an increased number of copies of a gene sequence, is seen in neuroblastoma (MYCN gene) (Fig. 14.5) and breast cancer (ERBB2 gene). Aneuploidy, a condition where the number of chromosomes in the nucleus of a cell is not an exact multiple of the haploid state, is seen in many cancers and results from errors of the mitotic checkpoint. Aneuploidy has been demonstrated in orbital rhabdomyosarcoma and ocular adnexal lymphoma.49,50
Role of micro-RNAs (miRNAs)
miRNAs are noncoding, single-stranded RNAs that function in RNA silencing and posttranslational expression of genes. miRNAs are thus negative regulators of genes. They participate in cell transformation either by causing overexpression of oncogenes or by reducing the expression of tumor suppressor genes (Fig. 14.6). Downregulation or deletion of miRNAs resulting in overexpression of Bcl2 is seen in some leukemias and lymphomas. Loss of miRNAs has also been shown to occur in mantle cell lymphoma, multiple myeloma, and prostate cancer.51

Cancer Epigenetics
Epigenetics refers to the study of heritability in gene expression that occurs independent of changes in the primary DNA sequence. These changes acquired during differentiation are maintained in a stable fashion during multiple cell divisions.52 This heritability is mediated by epigenetic modifications such as methylation of cytosine bases in DNA, posttranslational modifications of histone proteins, and positioning of nucleosomes along the DNA.53 Failure of the proper maintenance of heritable epigenetic marks can result in inappropriate activation or inhibition of various signaling pathways and lead to disease states such as cancer.53,54 Promoter hypermethylation of the RB1, HIC1, HIN1, and CDX-1 tumor suppressor genes has been observed in orbital rhabdomyosarcoma.55 Promoter hypermethylation p16, ECAD, and MT1G tumor suppressor genes have been observed in ocular adnexal lymphoma.56,57 One of these studies in a South Korean population also suggested the possible role of C. psittaci in triggering aberrant DNA hypermethylation of these genes.57 E-cadherin promoter hypermethylation has been observed in sebaceous eyelid carcinoma and has been associated with a poor prognosis.58 Loss of histone acetylation mediated by histone deacetylases (HDACs) result in gene repression. In a recent clinical trial, partial response to HDAC inhibitors has been reported in adenoid cystic carcinoma of the lacrimal gland, suggesting a possible role of HDACs in the development this tumor.59
Molecular Pathogenesis of Cancer
Cell transformation in cancer is a multistep and complex process (Fig. 14.5). Four types of regulatory genes participate in normal cell growth: proto-oncogenes (growth-promoting genes), which encode for the cell proliferation pathway; tumor suppressor genes, which are growth inhibitory genes; apoptosis-regulatory genes, which control the programmed cell death; and DNA-repair genes, which regulate the repair of DNA occurring during mitoses. The major hallmarks of cancer at the genetic level are discussed in the following sections.
Self-Sufficiency in Growth Signals
Under normal physiologic situations, a growth receptor binds to its specific receptor on the cell membrane, leading to transient and limited activation of the receptor, which, in turn, activates signal transducing proteins. These signals are transmitted across the cytosol to the nucleus by signal transduction molecules, with resultant induction and activation of nuclear regulatory factors that initiate and regulate DNA transcription and the eventual entry and progression of the cell into the cell cycle.
Growth Factors
Some cancer cells acquire growth self-sufficiency by acquiring the ability to synthesize the same growth factors to which they are responsive.47 Although benign in nature, orbital lymphatic malformations have been shown to express growth factor receptors, suggesting a possible therapeutic role of their inhibitors.60 A similar mechanism is present in some sarcomas that synthesize transforming growth factor-alpha (TGF-α) and its receptor.47 TGF-α promotes angiogenesis by inducing vascular endothelial growth factor (VEGF). Binding of TGF-α to epidermal growth factor receptor (EGFR) activates downstream signaling pathways, and this leads to cell proliferation and eventual neoplasia.
Growth Factor Receptors
Mutated growth factor receptors promote cell proliferation with minimal or no binding to growth factors. Examples of these are EGFR overexpression in orbital and periocular basal cell and SCCs.61
Signal-Transducing Proteins
Signal-transducing proteins facilitate the binding of growth factors to their nuclear targets. Mutations in genes encoding components of signaling pathways result in cancer growth autonomy. RAS, a member of the small G proteins, is the most commonly mutated oncogene in human cancers.47 The inactive RAS is guanosine diphosphate (GDP) bound. Growth factors result in exchange of GDP for guanosine triphosphate (GTP) and thus activate RAS. The intrinsic guanosine triphosphatase (GTPase) activity of RAS converts GTP to GDP, leading to inactivation of RAS. When mutated, RAS remains unaffected by GTPase and continues to promote cell proliferation. N-ras mutation has been demonstrated in orbital rhabdomyosarcoma.62 N-ras and k-ras mutations are seen in acute myeloid leukemias and may be seen in orbital granulocytic sarcoma.63
Nuclear Transcription Factors
Of various nuclear regulatory transcription proteins, the MYC gene located on chromosome 8 is the most important. In physiologic state, the MYC protein binds to DNA and regulates cell cycle by transcriptional activation. Its levels fall as soon as the cell enters the cell cycle. Persistence or overexpression of the MYC oncoprotein is seen in several human cancers, resulting in autonomous cell proliferation.64–66 The MYC protein can activate or repress the transcription of other genes. CDKs are activated by MYC, whereas CDK inhibitors are repressed by MYC. Thus, dysregulation of MYC promotes tumorigenesis by increasing the expression of genes that promote progression through the cell cycle and repressing genes that slow or prevent progression through the cell cycle.47 MYC dysregulation has been shown to occur in ocular adnexal lymphoma.67
Cell Cycle Regulatory Proteins
The cell cycle is under the control of cyclins, CDKs, and CDK inhibitors, as shown in Fig. 14.7. Complexes of cyclins with CDKs drive the cell cycle by phosphorylating various substrates. CDKs are controlled by inhibitors; mutations in genes encoding cyclins, CDKs and CDK inhibitors result in uncontrolled cell cycle progression. Mutations of cyclin D (translocation) are seen in mantle cell lymphoma and CDK4 (gene amplification) in malignant melanoma.68,69

Insensitivity to Growth Inhibitory Signals
Tumor suppressor genes provide the “brakes” during cell growth. Any mutation in these genes acts to remove the braking effect and paves the way for uncontrolled cell proliferation. Major tumor suppressor genes in the human cell cycle are RB, TP53, and transforming growth factor-beta (TGF-β). The RB gene located on 13q14 was the first tumor suppressor gene to be discovered and is considered a prototype for such a group of genes. It encodes for pRB, a nuclear transcription protein. The active form of RB blocks cell division by binding to transcription factor E2F, thus inhibiting the transcription of cell cycle–related genes and eventually inhibiting the cell cycle at the G1-S phase. The inactive form of the RB gene results from hyperphosphorylation by CDKs, which removes the “brake” and promotes the passage of cell through the G1-S phase and leads to cell proliferation (Fig. 14.8). This activity of CDKs is blocked by TGF-β through activation of p16. The RB gene mutations are seen in several human tumors, including retinoblastoma.47 For retinoblastoma to develop, both normal alleles of the RB locus need to be inactivated. In familial cases, children inherit one abnormal copy of the RB gene. These children develop retinoblastoma when the other normal copy is lost as a result of somatic mutation (Fig. 14.9). In sporadic cases, both normal alleles are lost as a result of somatic mutations (Fig. 14.10).



TP53, also known as the guardian of the genome, is one of the most commonly mutated genes. p53, the protein encoded by TP53, serves two important functions in the cell cycle: inhibition of cyclins and CDKs, thus preventing cells from entering G1, and promotes apoptosis by activating the BAX gene. With homozygous loss of the TP53 gene, the DNA damage is unrepaired, and such genetically impaired cells continue to proliferate. p53 mutations are seen in eyelid sebaceous carcinoma and orbital rhabdomyosarcoma.70,71 Further, p53 expression has been correlated with differentiation and proliferation in orbital rhabdomyosarcoma, which suggests its role as a potential prognostic indicator in this tumor.71
TGF-β, after binding to the TGF-β receptor, inhibits cell proliferation by activating CDK inhibitors and suppresses growth promoting genes such as MYC, cyclins, and CDKs. Mutations of the TGF-β gene cause loss of the inhibitory effect and promotes cell growth. The TGF-β/SMAD signaling pathway has been implicated in the pathogenesis of basal cell carcinoma.72
Other tumor suppressor genes described in orbital tumors include the von Hippel Lindau (VHL) and neurofibromatosis (NF) genes.
Evasion of Apoptotic Cell Death
Abnormal cells in cancer accumulate not only as a result of activation of oncogenes or inactivation of tumor suppressor genes but also because of mutations in genes regulating apoptosis. Apoptosis, or programmed cell death, is a genetically determined process. The apoptotic pathway has upstream regulators and downstream effectors. The regulators act through the extrinsic (death receptor) or intrinsic (mitochondrial) pathways. As shown in Fig. 14.11, stimulation of extrinsic and intrinsic pathways results in activation of caspases 8 and 9, respectively, which, in turn, initiates a proteolytic cascade leading to apoptosis. Disrupted apoptosis is seen in several human cancers. In BL, t(14;18) (q32;q21) results in overexpression of the Bcl2 protein, which protects lymphocytes from apoptosis, allowing genetically damaged lymphocytes to survive longer and proliferate.73

Limitless Replicative Potential, Telomeres, and Telomerase
After a certain number of mitoses (cell doubling), human cells cease to divide and instead become senescent. This property of cellular aging has been attributed to shortening of telomeres after every cell division. Short telomeres are recognized by the DNA repair mechanisms, leading to cell cycle arrest mediated by the TP53 and RB genes. Mutations of these genes result in disabling of these checkpoints and activation of the nonhomologous end-joining pathway to join the shortened ends of two chromosomes with the effect of saving the cell. An inappropriately activated repair system results in dicentric chromosomes that are pulled apart at anaphase, resulting in new double-stranded DNA breaks. The repeated bridge–fusion–breakage cycle eventually produces a mitotic catastrophe and massive apoptosis. Reactivation of telomerase results in cessation of the bridge–fusion–breakage cycle, and the cell avoids death. Upregulation of telomerase with resultant maintenance of telomere length is seen in eyelid sebaceous carcinoma.74,75
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


