Vascular malformations
Venous
Lymphatic
Capillary
Arterial and arteriovenous
Mixed (combined)
Vascular neoplasms
Infantile hemangioma (IH)
Congenital hemangioma (CH)
Rapidly involuting (RICH)
Non-involuting (NICH)
Others
Kaposiform hemangioendothelioma (KHE)
Tufted angioma (TA)
Lobulated capillary hemangiomas (LCH) (pyogenic granuloma)a
The most common vascular anomalies of the neonatal airway are infantile hemangiomas, lymphatic malformations, and venous malformations. Infantile hemangiomas, which are among the vascular tumors, have a predilection for the subglottic airway. In contrast, lymphatic and venous vascular malformations are typically pharyngeal and supraglottic and cause obstruction in higher regions of the airway [4].
Infantile Hemangioma of the Airway
Infantile hemangiomas (IHs) are benign tumors of the vascular endothelium. The most common tumors of infancy, IHs have the unique ability to involute after a period of rapid proliferation. Most IHs involve the skin, while only a small percentage involve the mucosa or the viscera.
IHs of the neonatal airway are relatively uncommon. Also known as “subglottic” hemangiomas, these lesions have a tendency to affect the narrowest portion of the pediatric airway, and therefore present with symptoms of stridor and cough. Growth of the tumor may result in airway compromise and severe respiratory distress.
Epidemiology
The incidence of IH in general has been estimated at 4–5 % based on retrospective and cross-sectional data [5], although other studies suggest rates of 1–3 % among newborns [6, 7], and 2.6–9.9 % among older children. [8, 9]. IHs occur more commonly among female infants at rate of about 3:1 [9, 10], and most studies report a significantly higher incidence in Caucasian infants [9–11]. The incidence of IH is increased among preterm infants, affecting 22–30 % of babies weighing less than 1 kg [10, 12]. Low birth weight has been implicated as the causal reason, with a 25 % increase in risk of IH development for every 500 g reduction in birth weight [13].
In a 1967 series, IHs of the airway accounted for 1.5 % of congenital laryngeal anomalies [14]. However, the actual incidence of IHs has not been determined in any formal study. An analysis of the 37-hospital Pediatric Health Information System (PHIS) database over the 5-year period from 2001 to 2005 found that of 2,890 admissions for a primary or secondary diagnosis of IH, 337 (12 %) underwent an airway procedure during at least one admission [15]. Thus, on average, pediatric hospitals in this cohort likely treated fewer than three symptomatic airway IHs each year. As with IH in general, airway IH of the subglottis have been reported more frequently in females, but with a 2:1 female to male preponderance [16–20]. It is unknown if the other risk factors associated with IH in general apply equally to the subset of IHs of the airway.
Etiology and Pathogenesis
The etiology and pathogenesis of IH remain theories that are incompletely proven. The endothelial progenitor cell theory suggests that IHs develop from clonal expansion of circulating endothelial progenitor cells (EPCs), resulting in vasculogenesis, or the de novo formation of new blood vessels. [21, 22]. The proposed stimulus for division of EPCs is a somatic mutation or abnormal signals from local tissues. The theory of placental origin suggests that fetal progenitor cells arise from disruption of the placenta during gestation or birth. This concept derived from research demonstrating that molecular markers unique to placental tissue were also present in IHs [23–25]. Clinical evidence for this theory is suggested by studies demonstrating an increased incidence of IH in association with chorionic villus sampling, placenta previa, and preeclampsia.
A unifying theory suggests that circulating EPCs find their way to certain locations that provide conditions favorable for growth into placenta-like tissues. In such tissues as the skin and liver, progenitor cells may encounter cellular signals and local tissue factors that stimulate their development. Such factors may include angiogenic and vasculogenic factors within the IH [26–28]. It is also theorized that disturbances causing placental hypoxia trigger a vascular response that increases the likelihood of IH [29–31]. Such disturbances include maternal chorionic villus sampling (CVS) or amniocentesis [[10, 32, 33], and placental anomalies such as retroplacental hematoma, infarction, and dilated vascular communications [29]. In utero hypoxia is also the most common cause of low birth weight and may explain the association seen with premature delivery.
Clinical Presentation
Although considered congenital lesions, airway IHs are not generally symptomatic at birth. However, like cutaneous IH, they typically exhibit a characteristic life cycle consisting of proliferation and involution. Proliferation occurs during early infancy, while gradual spontaneous involution or regression starts by 1 year of age [34–41]. Symptoms usually manifest during the early proliferative phase; accordingly, some airway IHs will become symptomatic during the neonatal period. 80–90 % of affected babies will present within the first 6 months with a mean age of 3.6 months at diagnosis [16, 42]. Symptoms are often present for several weeks before a definitive diagnosis is made. An intermediate period between proliferation and involution during mid-to-late infancy, often referred to as the “plateau” phase, more likely represents a period of temporary balance between individual cells that are proliferating and those undergoing involution and apoptosis [34–41]. The process of involution takes several years and varies in duration.
Most symptomatic airway IHs involve the subglottic larynx and, as a result, authors have traditionally referred to these lesions as “subglottic hemangiomas.” In fact, many such IHs have been demonstrated to have transglottic or paratracheal extension [43]. Nevertheless, their associated symptom complex is consistent, including biphasic stridor and barky cough, usually in the absence of dysphonia. The symptoms are often mistaken for those of infectious or inflammatory croup, especially since the symptoms typically worsen in the presence of upper respiratory illness. Furthermore, IHs respond to many of the same treatments used for croup, including racemic epinephrine and nebulized and systemic steroids. As a result, recurrent croup-like symptoms and gradual worsening of stridor during early infancy are suggestive of airway IH and should be investigated further. Swallowing is usually normal; however feeding may be affected as the infant tries to coordinate sucking with breathing.
About one half of infants diagnosed with an airway IH also will have a cutaneous IH, although only 1–2 % of children with cutaneous IHs also have airway IHs [17, 44]. Even in asymptomatic children, the presence of cutaneous IH in the “beard distribution” (parotid area, lips, chin, neck) may be a predictor of IH in the airway (Fig. 1) [45]. Such cases likely represent “segmental” IHs [43, 46].
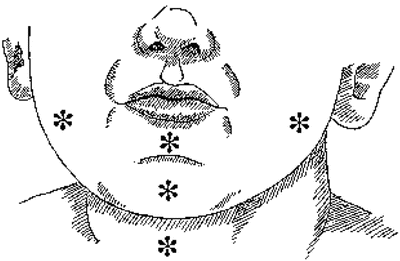
Fig. 1
Subsites of IHs in beard distribution, denoted by asterisks (Reprinted from [45], copyright © 1997, with permission from Elsevier, Ltd)
Once the proliferative phase is completed (9–12 months of age), respiratory symptoms tend to slowly resolve. Although the lesion itself may not involute completely or even significantly for many months, growth of the child over time allows the airway to better accommodate the IH, resulting in diminished stridor and cough.
Diagnosis
Diagnosis of airway IH begins with a high index of suspicion, since many congenital and inflammatory processes of the airway can mimic IH during the early months of infancy. The presence of cutaneous lesions and the persistence or recurrence of croup-like symptoms are important clues. When airway IH is suspected, the diagnosis may be confirmed by performing an imaging study or proceeding directly to laryngoscopy and bronchoscopy. In most cases, the uncertainty of the diagnosis or the severity of symptoms necessitates a trip to the operating room, where the pathology may be visualized directly and treated surgically if necessary. This approach avoids the risks associated with radiation exposure during infancy. Advocates of imaging, conversely, prefer to make a presumptive diagnosis based on flexible airway endoscopy, and confirm the diagnosis and assess the extent of the lesion by CT (computerized tomography) scan with angiography (“CT angio”) [47]. This approach avoids the need for general anesthesia, especially given the recent trend to treat such lesions initially with medical therapy rather than surgery.
When endoscopy is performed, symptomatic airway IHs are usually found to involve the subglottis, but bulk and blush may be present at adjacent and distant sites as well. The bulky lesion is smooth, submucosal, compressible, and pink or blue in color (Fig. 2). A left-sided predominance has been reported, but IHs may be bilateral, circumferential, or multiple. Biopsy is not usually necessary to establish the diagnosis; however when the diagnosis is in doubt, specimens from true IHs will stain positively for GLUT1 (glucose transporter protein isoform 1) [24, 48]. The most useful and widely used immunohistochemical marker for the diagnosis of IH, GLUT1 is strongly expressed by IH endothelial cells and not by other benign vascular anomalies, and has been validated for the identification of IHs of the airway [49].
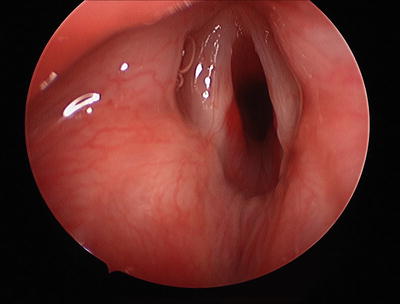
Fig. 2
Hemangioma on posterior wall of glottic and subglottis
On CT imaging, IHs will demonstrate an intensely staining, well-circumscribed mass with lobular architecture. MRI, although often diagnostic, is less advantageous since the duration of the study generally requires general anesthesia, often with endotracheal intubation. Fluoroscopy or anterior-posterior radiographs of the neck may demonstrate an asymmetric subglottic narrowing; however these studies do not definitively establish the diagnosis.
A staging system for airway IHs has been proposed by Perkins et al. [47]. IH stage is determined from CT angiography based on location, percentage of laryngeal airway obstruction, and estimated total volume of the IH (Table 2).
Table 2
Proposed airway IH staging system and treatment protocol
Stagea | Unilateral airway hemangioma | Circumferential or bilateral airway hemangioma | Percent laryngeal airway obstruction | CTA or MRI extralaryngeal hemangioma volume (mm3) |
|---|---|---|---|---|
1 | Yes | No | ≤50 | <4,000 |
2 | Yes | Yes | >50–90 | 4,001–10,000 |
3 | No | Yes | >90 | >10,000 |
Management
Once the diagnosis of airway IH has been established, the need for and type of intervention is determined by several factors, including the degree of airway obstruction, the extent of extralaryngeal IH, the location of the patient at the time of diagnosis, the experience of the treating physician, and the preferences of the caretaker.
Since involution is the ultimate fate of nearly all infantile IHs, “watchful waiting” is reasonable in cases involving minimal symptomatology. For those patients with symptomatic IHs whose caretakers prefer no intervention for the lesion itself, tracheotomy with observation is highly successful [18]. This approach, however, requires a high level of maintenance to avoid tube occlusion, accidental decannulation, and exposure of the airway to water over a long period of time. As a result, in most cases, some sort of intervention for the IH itself is more desirable.
Pharmacotherapy
In a landmark 2008 publication by Léauté-Labrèze et al., the authors described their serendipitous observation that involution of IHs may be accelerated with the administration of propranolol [50]. At doses of 2–3 mg/kg/day used to treat cardiac complications of their IHs, two children experienced marked and rapid involution of their IHs. These results were replicated in a case series reported by the same authors [51] and in publications by a number of other investigators [52–62].
Several mechanisms have been proposed to explain the mechanism by which propranolol inhibits IH growth, including vasoconstriction due to decreased release of nitric oxide, blocking of proangiogenic signals (vascular endothelial growth factor, basic fibroblast growth factor, matrix metalloproteinases 2 and 9), and induction of apoptosis in proliferating endothelial cells [51, 62–64]. It has also been suggested that propranolol may prevent the differentiation of IH stem cells into endothelial cells or pericytes [65], or that it may hasten the differentiation of progenitor cells into adipocytes [51].
Several clinicians have reported success using propranolol in the management of airway IHs [66–76], but response is not universal [77]. The pretreatment assessment, optimal dose, and appropriate duration of therapy vary considerably in the literature. After considering contraindications such as cardiogenic shock, sinus bradycardia, hypotension, heart block greater than first-degree, heart failure, bronchial asthma, and known hypersensitivity to the drug, most clinicians will perform a cardiac evaluation, including at least an examination and a pretreatment electrocardiogram. In most series, the drug is started at a dose of 1 mg/kg/day divided two to three times a day, and then increased over several days to a week to 2–3 mg/kg/day. Heart rate and blood pressure are checked each hour for the first 2–3 h after the initial dose and with each dosage increase. The drug is administered throughout most of the first year of life. Although protocols for propranolol initiation do appear in the otolaryngology literature [78, 79], a consensus multidisciplinary protocol has also been published [62].
Propranolol has a well-established safety profile based on years of use at higher doses for control of high blood pressure and cardiac pathology. Potential side effects and complications include sinus bradycardia, hypotension, reactive airways, hypoglycemia with secondary seizures, diarrhea, and cool extremities. Patients should be monitored for evidence of bradycardia, hypotension and/or hypoglycemia [62, 65, 80]. However, such complications are reported rarely, and it has been suggested that propranolol should become the standard for initial management of all airway IHs [81].
Although propranolol has largely supplanted systemic corticosteroids as first line pharmacotherapy, the latter are occasionally useful in refractory cases. Steroid medications inhibit growth of the lesion during the proliferative phase, losing their effectiveness once involution begins [82]. Doses of prednisolone at 2–3 mg/kg/day are generally necessary to control growth of the mass and should be maintained for 1–2 weeks before starting a 2–4 week taper. Response rates reported in the literature vary between 30 and 93 %, although there is little consistency among dosing regimens [82, 83]. Long-term management on steroids carries a significant risk of complications, including gastroesophageal reflux, gastritis, immune suppression, cushingoid changes, hyperglycemia and glycosuria, hypertension, fluid and electrolyte disturbances, and growth retardation [82]. IH patients on maintenance steroids should concomitantly receive courses of H2 receptor blockers and trimethoprim-sulfamethoxasole as prophylaxis against gastritis and pneumocystis carinii infection, respectively. Live vaccinations should also be avoided while a child is taking high-dose steroids.
Interferon α-2A has been used with success in treating IHs, but should only be considered when all other traditional modalities fail [84–87]. Potential side effects are associated with this therapy, including fever, myalgia, transient elevation of hepatic transaminase levels, transient neutropenia, anemia, and spastic diplegia [88, 89].
Surgical Intervention
Indications for surgical management of airway IH have become few since the efficacy of propranolol was established. Surgery is a reasonable consideration when (1) the obstruction found at operative endoscopy is severe and will likely require a lengthy intubation while medical therapy is initiated, and (2) the patient remains severely symptomatic despite an adequate trial of medical therapy.
Operative intervention may include intralesional injection of corticosteroids and/or partial ablation of the IH, or complete surgical excision the portion of the lesion within the airway. In most cases, the patients will remain on medical therapy postoperatively to reduce the likelihood of recurrence. This is of particular importance in “segmental” airway lesions with known extension outside of the airway.
Intralesional steroids should be considered for patients whose IHs have necessitated a trip to the operating room for endoscopy or endoscopic resection. While repeated injections are usually necessary as single modality therapy [90], these medications may be effective adjuvant therapy for patients whose lesions are being observed, treated pharmacologically, or partially resected. In most cases, triamcinolone 40 mg/cm3 is administered at a dose of 3–5 mg/kg either alone or supplemented by betamethasone 6 mg/cm3 dosed at 0.5–1.0 mg/kg (Fig. 3) [82]. Total volume delivered may be limited by the size of the lesion, and care must be taken to avoid depositing the steroid medication deep enough to affect the underlying cartilage. Patients will usually require at least overnight intubation due to the increased volume of the lesion after injection. Cure rates of 77–87 % using intralesional steroids have been reported [42, 90].
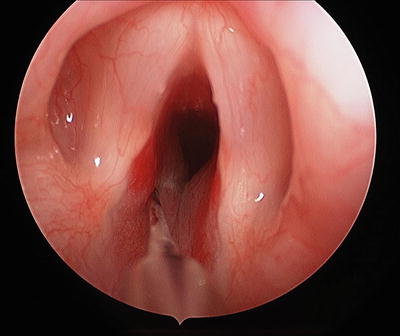
Fig. 3
Triamcinolone/betametasone steroid injection with needle in left side of subglottic hemangioma
Airway IHs causing focal obstruction may be addressed surgically by a subtotal endoscopic approach using a microscope or telescope, or by total excision through an open approach, dividing the thyroid and cricoid cartilages in the midline. Subtotal resection, more often than total excision, carries the risk of growth of the residual lesion during the proliferative phase, potentially resulting in additional surgical procedures unless combined with pharmacologic therapy. Endoscopic excision is usually performed using an apneic anesthesia technique, intermittently interrupting the surgery for reinsertion of the tube and ventilation of the patient. Alternatively, the procedure may be performed under spontaneous ventilation with anesthetic insufflation or Venturi jet ventilation.
The laser has been the most popular endoscopic surgical modality [42], with the carbon dioxide (CO2) [17, 19, 44, 91, 92] potassium titanyl phosphate (KTP) [93, 94], and neodymium:yttrium-aluminum-garnet (Nd:YAG) [95, 96] lasers all demonstrating some effectiveness. All of these lasers are currently available for airway use through fiber delivery systems, however only CO2 is used by direct beam. CO2 lasers are preferentially absorbed by water, while KTP and Nd:YAG lasers take advantage of absorption peaks that approximate those of hemoglobin and are thought to penetrate more deeply. However, all of these lasers cause destruction by ablation rather than selective photothermolysis. As a result, in addition to the risk of recurrence, laser treatment carries a risk of subglottic stenosis of 5–25 % that is likely greatest with deeper resections and in cases of bilateral or circumferential disease [19, 44, 89, 97]. Debulking of the lesion using rotary powered instrumentation (microdebrider or “shaver”) has also been reported [98, 99]. Postoperatively, patients are observed in an intensive care setting. Some clinicians recommend face tent humidification to prevent airway obstruction due to eschar formation.
Although the first open surgical excision of a focally obstructing airway IH was reported in 1949, the procedure did not gain popularity until the 1990s, after complications of laser therapy became increasingly apparent [100–105]. Over the 15 years prior to the discovery of effects of propranolol, open resection appeared to be emerging as the intervention of choice for airway IHs. The procedure is of greatest advantage in patients with bilateral or circumferential lesions that may otherwise been at risk for postoperative stenosis, recurrence, or tracheotomy. However, open surgical excision may be more difficult in cases involving significant extra-laryngeal extension, and the procedure may potentially result in some degree of dysphonia.
After initial intubation through the obstructed portion of the airway, the lesion is approached through the anterior neck via laryngofissure. After the tube has been relocated to the inferior aspect of the incision, the IH is removed submucosally under the operating microscope. At the conclusion of the dissection, the patient is intubated; in some cases a thyroid cartilage graft may be placed to enlarge the subglottic laryngeal framework. After the neck is closed, the patient is transported to the intensive care unit where intubation is maintained for 3–7 days.
Multidisciplinary Considerations
Management of airway IH requires good communication among pediatric specialists, both in terms of diagnosis of the lesion as well as management after the diagnosis has been made. It is estimated that some 50 % of patients with airway IHs have cutaneous lesions as well, many with problematic segmental involvement in the “beard” distribution. In addition, there is an important association of airway IH with PHACE(S) syndrome, including Posterior fossa defects, Hemangiomas, cerebrovascular Arterial anomalies, Cardiovascular anomalies including Coarctation of the aorta, Eye anomalies, and Sternal cleft and/or Supraumbilical raphe (OMIM 606519) [106]. In one study of 23 PHACE patients, 52 % were found to have airway IH, many of whom required treatment for stridor [107]. Another study found eight patients (42 %) meeting the criteria for PHACE among 17 with large “beard” distribution IHs and airway IHs [108]. In such cases, early consultations with pediatric dermatology and genetics are advisable.
During diagnostic endoscopy, as with all lesions of the airway, moment-to-moment communication between the otolaryngologist and the anesthesiologist is critical to successful management. Since in most cases a subglottic lesion is predictable based on symptoms, the two clinicians should have a game plan in place for intubation, management of the airway during any intervention, and emergency contingencies, and the two should agree on a plan for airway management in the immediate postoperative period.
Postoperatively, the otolaryngologist must continue to coordinate the airway management plan with the pediatric intensivists and hospitalists that will administer care to patients with airway IH while they recover from endoscopy and start on medical therapy. Such care includes the duration of intubation (if any), management of medical therapy for the IH and for reflux if indicated, and use of adjunctive racemic epinephrine and/or steroids to control exacerbations of stridor. In most cases, consultation with a pediatric cardiologist is advisable prior to starting propranolol therapy while the child is still hospitalized.
Future Considerations
There are still several important unanswered questions in the diagnosis and management of IH of the airway. One key issue in pathogenesis is the predilection of IH to involve the subglottis as opposed to other areas of the airway. As the etiology of IH in general becomes clearer, it may be possible to theorize plausible reasons for this clinical observation. It is also not yet clear if there is some subset of patients with a presentation of cutaneous IH that should undergo endoscopy prior to the development of symptomatic airway obstruction. Finally, in terms of therapeutic intervention, propranolol therapy is a relatively new tool in the management of airway IH, and additional research will be required to determine optimal administration, including dosage, frequency, and duration of therapy.
Vascular Malformations of the Neonatal Airway
Vascular malformations are congenital disorders of vasculogenesis with ectatic vascular networks and the propensity for relentless expansion and recruitment of new vessels. Unlike hemangiomas, vascular malformations do not experience a cycle of growth and involution. They are present at birth and grow commensurate with the child until periods of rapid expansion lead to functional and aesthetic compromise. Trauma, infection, and hormonal fluctuation have been associated with acute growth of these vascular anomalies [109, 110].
Vascular malformations may be composed of any blood vessel type and they are named accordingly, including capillary malformations (port wine stains), venous malformations, lymphatic malformations, and arteriovenous malformations. The most common vascular malformations affecting the neonatal airway are lymphatic and venous malformations [111–113]. Relative to subglottic hemangiomas and other disorders of the newborn airway, these malformations are rare. Although they may occur in isolation, neonatal vascular malformations of the airway are more commonly associated with diffuse cervicofacial lesions [114, 115]. Diffuse involvement of the head and neck will commonly affect the aerodigestive tract mucosa to include the oral cavity, tongue, hypopharynx and larynx (Fig. 4) [113]. Vascular malformations typically experience slow growth but airway involvement may necessitate urgent intervention, especially in the setting of acute or chronic obstruction.
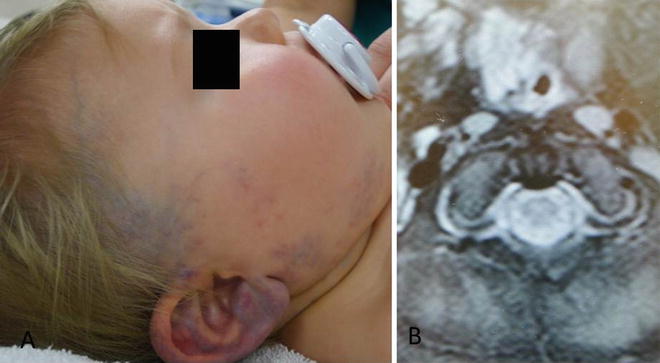
Fig. 4
Infant with venous malformation of facial soft tissue and larynx. The malformation is characterized by blue cutaneous staining (left) and enhancing lesion of supraglottis on MRI (right)
Lymphatic Malformations
Epidemiology
Lymphatic malformations are believed to occur in 1 in 500 live births and comprise about 12 % of vascular cases referred to a vascular center [116]. Within the head and neck, lymphatic malformations occur at a rate of between 1 in 2,000 and 1 in 4,000 [117]. Fifty percent of cases present after birth and 80–90 % by 2 years of age. There does not appear to be a gender or race predilection for LMs.
Etiology and Pathogenesis
Lymphatic malformations (LMs) are slow flow vascular malformations composed of dilated ectatic lymphatic channels. They are thought to arise from disruptions in embryonic lymphangiogenesis but their etiology and pathogenesis remain unclear. Lymphatic malformations are present at birth but may not be detectible until later in life. The majority of LMs occur in the head and neck, making them the second most common congenital mass found at these sites [118, 119]. Exacerbated by infection, cervicofacial LMs may expand in infancy or early childhood when the rate of ear and upper respiratory infections are at their peak. Diffuse lesions are more likely to progress than those that are localized. If left untreated, the likelihood of progression is significantly higher in adolescence than childhood [120]. The earlier intervention may thus be warranted in many cases.
Clinical Presentation
Based upon their response to treatment, LMs have been categorized by the size of the fluid filled lymphatic cysts present. In particular, cysts greater than 1–2 cm in diameter are considered macrocysts, while microcysts are those 1–2 cm in diameter or less [121]. While LMs may be predominantly macrocystic or microcystic, most LMs of the head and neck region are mixed lesions. Venous malformations may also be present within extensive cervicofacial LMs.
Macrocystic and microcystic LMs have different clinical courses. Macrocystic LMs grow by accumulated lymphatic fluid and expansion of the cysts into areas of least resistance. They have a tendency to encompass and compress surrounding architecture. In the neonate, they can affect the larynx and trachea by direct or indirect compression of these structures. Stridor, grunting, or symptoms consistent with obstructive sleep apnea may be present when the larynx, trachea, or pharynx are involved, respectively. With rapid expansion or internal bleeding, macrocystic LMs may cause acute compromise of vital structures. Chronic expansion in the head and neck frequently leads to upper aerodigestive symptoms such as dysphagia and gradual airway obstruction, especially during recumbence.
Unlike macrocystic LMs, microcystic LMs are infiltrative in nature. In this condition, small microcystic vesicles penetrate soft tissue including skin, muscle, and mucosa [122, 123]. Non-descript soft tissue swelling and the emergence of weeping vesicles in the skin or mucosa often note its presence. Isolated disease commonly occurs in the tongue, upper lip, and larynx (Fig. 5). When associated with mixed cervicofacial lesions, microcysts in the tongue and larynx can cause acute life-threatening airway obstruction upon delivery, shortly after birth, or during periods of acute infection and swelling (Fig. 6) [124].
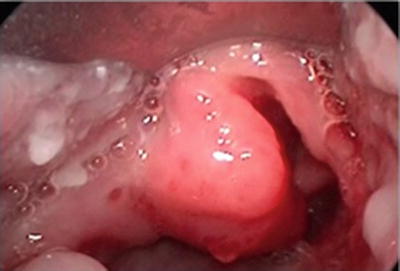
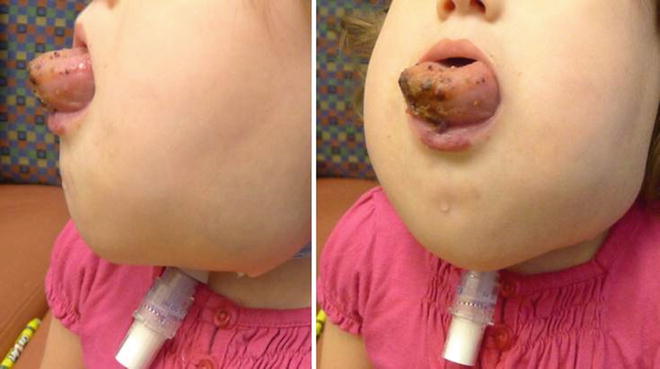
Fig. 5
Isolated lymphatic malformation of the supraglottic larynx. Lymphatic vesicles are seen on the left pharyngeal wall and vallecula
Fig. 6
Cervicofacial microcystic lymphatic malformation with multiple levels of airway obstruction leading to early tracheostomy
Isolated microcystic LMs rarely cause respiratory obstruction in the newborn period. However, in the presence of massive cervicofacial disease of mixed or microcystic LM, rapid airway compromise may occur, especially during periods of acute edema [124]. Upper airway obstruction is exacerbated by infiltrating disease of the tongue and floor of mouth (Fig. 6) [123, 125–127]. Laryngeal involvement will exacerbate symptoms.
Diagnosis
Airway endoscopy with office-based flexible laryngoscopy, followed by rigid microlaryngoscopy and bronchoscopy, will provide best evidence of the extent of obstruction and its effect on the airway. Distinct margins of macrocystic LMs may also be detected on physical exam and on magnetic resonance imaging (MRI), the diagnostic study of choice for LMs. An MRI of the head and neck will demonstrate the depth of disease and differentiate macrocystic LM from more common conditions of the pediatric head and neck. Hyperintense lesion on T2 weighted images and air-fluid levels within the lesion are hallmark MRI findings for macrocystic LM [128]. Frequently mixed lesions are discovered. Microcystic and macrocystic lymphatic malformations may coexist with deep neck space venous malformations which will also be evident on MRI.
Management and Multidisciplinary Considerations
Except in rare cases, LMs ultimately need intervention to control symptoms associated with their accumulated lifetime growth. Timing of intervention for LMs in infants is dependent upon the acuity of symptoms and dictated by the presence, or risk of, acute and chronic upper aerodigestive dysfunction or pain due to inflammation. Acute airway obstruction, sleep apnea, and dysphagia are the most common reasons for intervention in these patients [113, 118, 126]. Sclerotherapy and surgery constitute the therapeutic approach to lymphatic malformations and is typically conducted in a multidisciplinary fashion. However, symptom control may be best achieved acutely with oral steroids and antibiotics that reduce the inflammation, lymph accumulation, and expansion of LMs.
Macrocystic Disease
A small percentage of isolated macrocystic LMs have been reported to spontaneously resolve [129, 130]. Because these isolated lesions usually occur in the neck, they rarely cause significant dysfunction in the newborn and can be observed into early childhood before treatment is considered.
Most macrocystic LMs compressing the neonatal airway require treatment. In acute situations, needle aspiration of the responsible cyst or cysts may alleviate the airway compromise until more definitive management with either surgical excision or sclerotherapy is possible. Sclerotherapy is the direct injection of agents that cause epithelial damage and subsequent disruption of the LM channels. Various materials have been employed with equal success including doxycycline, ethanol, sodium tetradodecyl, and OK-432, an immunostimulant derived from Group A streptococci [131–135]. With these therapies, dissolution occurs as the result of intravascular injury and luminal absorption by the body. Multiple treatment sessions, under radiographic fluoroscopy, may be required for significant reduction in volume using this technique. Postoperative edema with potentially worsening compression may occur following sclerotherapy. Expectant airway management with intubation or intensive care observation is necessary when treating compressive lesions of the neonatal airway. Risk to adjacent nerves ranges from 5 to 10 % with temporary dysfunction being more common than complete injury [134].
Surgical excision can provide immediate relief of airway obstruction caused by macrocystic cervical LMs. The risk of injury to important neurovascular structures in the neck is real but fortunately rare in experienced hands. Coexistent venous malformations may be found during surgical excision of deep neck LMs. Recurrence, persistent lymphatic drainage, and postoperative scarring often occur following excision [136]. These risks are minimized by removing as much disease as possible, placing perioperative drains, and hiding incisions in natural creases of the neck.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


