Fig. 1
The Benjamin and Inglis cleft classification
Clinical Presentation
Respiratory and feeding difficulties are the hallmark symptoms of LCs, though they are generally nonspecific. The clinical presentation and severity of symptoms correlate to the extent of the cleft, and the acuity of presentation may range from incidental diagnosis of an asymptomatic cleft to neonatal life-threatening respiratory distress.
Type I and some Type II LCs can be asymptomatic or present with stridor, persistent coughing or choking with feeds, regurgitation, hoarseness, failure to thrive, stridor, shortness of breath, and recurrent aspiration pneumonia. Type II LCs and Type III LTECs tend to present with the same symptoms but worse in frequency and severity. Cyanosis with feeds, chronic aspiration pneumonia, and increased airway obstruction with stridor may be seen; noninvasive airway support with positive pressure or oxygen supplementation may be required. Type IV LTECs are marked by early respiratory distress and require emergent intervention.
Over 50 % of patients with LCs have an associated syndromic or nonsyndromic congenital abnormality, and the severity of the cleft may correspond with the incidence of concurrent anomalies [3, 6, 21, 22]. The most frequently identified concurrent pathologies are related to the aerodigestive system and include tracheomalacia, tracheoesophageal fistula, laryngomalacia, and gastroesophageal reflux. Other reported malformations include gastrointestinal (esophageal atresia, microgastria, imperforate anus), genitourinary (hypospadias, kidney anomalies), craniofacial (Pierre Robin sequence, cleft lip with or without cleft palate, choanal atresia), and cardiac and cardiovascular disorders (ventricular septal defect, aorta anomalies) [3, 15, 23].
Laryngeal clefts have been identified as part of several well-described syndromes, including Opitz-Frias Syndrome (cleft lip, cleft palate, hypospadias, external ear anomalies, and hypertelorism) and Pallister-Hall syndrome (central neurologic system anomalies, imperforate anus, cardiac, pulmonary, renal, and distal extremity abnormalities). LCs may also be seen with CHARGE syndrome (coloboma, heart defects, atresia of the nasal choanae, retardation of growth, genitourinary defects, and ear defects), Down Syndrome, and the spectrum of 22q11 deletion syndromes. Additionally, LCs have been identified as part of the congenital VACTERL association (vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal fistulae, ear, renal, and limb anomalies).
Diagnosis
As the symptoms of a LC are nonspecific, the differential diagnosis is broad and includes laryngomalacia, tracheomalacia, tracheoesophageal fistula, gastroesophageal reflux, neuromuscular dysphagia, esophageal stricture, cricopharyngeal spasm, and unilateral or bilateral vocal cord paralysis. Even when the presentation is subtle, early definitive diagnosis is critical to an optimal outcome since chronic aspiration can lead to failure to thrive and chronic interstitial lung damage [22].
As with most pediatric airway pathology, endoscopic operative evaluation with microlaryngoscopy and rigid bronchoscopy is the mainstay of diagnosis and should be performed in any patient with persistent coughing, choking with feeds, stridor, respiratory distress, hoarseness, or recurrent aspiration pneumonia. The diagnosis of LC must be kept in mind during evaluation, as it can be easily missed when redundant mucosa prolapses into the cleft. The endoscopy is generally performed under general anesthesia with spontaneous ventilation, as endotracheal intubation would preclude an adequate exam. Several operative techniques have been described to examine the posterior laryngotracheal complex. Regardless of preferred technique, the interarytenoid space must be carefully examined and palpated to determine the presence and extent of a LC. Since flexible laryngoscopy and flexible bronchoscopy lack the required exposure and access for palpation, rigid endoscopy is the gold standard when evaluating for a LC (Fig. 2). Over one third of patients with a LC may also have a tracheoesophageal fistula; therefore, the initial identification of a cleft must not obviate the completion of a thorough bronchoscopic evaluation [6, 24]. The diagnosis of the Type I LC can be particularly challenging, as the symptoms are mild and the clefts subtle [8, 9, 21].
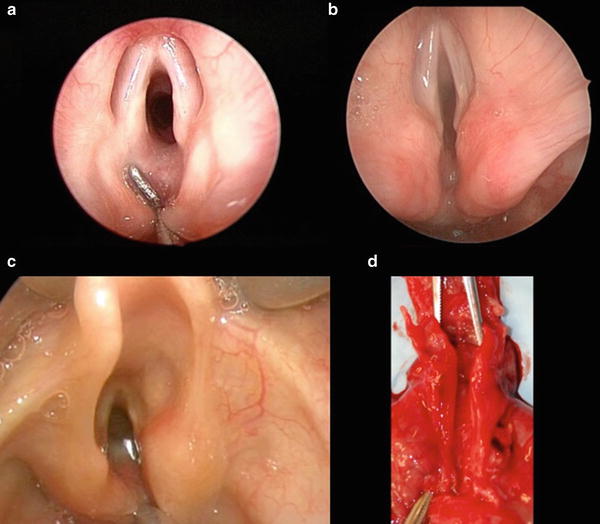
Fig. 2
(a) Type 1, (b) Type 2, (c) Type 3, (d) postmortem specimen of a Type 4 LTEC
Several ancillary tests have proven useful in conjunction with endoscopy. Flexible fiberoptic laryngoscopy with the patient awake should be performed to determine vocal cord function and the presence and severity of potential concurrent laryngomalacia. A modified barium swallow and FEES (fiberoptic endoscopic examination of swallowing) examination are valuable for diagnosing and quantifying aspiration. It should be noted, however, that aspiration is a nonspecific finding for a LC and that normal swallow studies do not rule out the diagnosis of a cleft. Chest radiography can demonstrate findings of aspiration pneumonia and the development of interstitial lung disease. Finally, flexible bronchoscopy and bronchoalveolar lavage for lipid laden macrophages may quantify the severity of aspiration and help to predict which patients will benefit most from early surgical therapy [5]. When a LC is identified, a systemic evaluation is required to rule out concurrent congenital anomalies, including genetic, cardiothoracic, genitourinary, gastrointestinal, vertebral, and neurologic assessment.
Management
The treatment of LCs varies based on the extent of the cleft and the severity of the symptoms, but the underlying management principles are consistent. Airway protection is always the immediate concern and must be addressed first. When required, respiratory support should be delivered by noninvasive means if possible, such as nasal continuous or biphasic positive airway pressure, rather than prolonged endotracheal intubation. Intubation may lead to additional trauma and inflammation of the planned surgical site. When tracheotomy is required for severe clefts or due to associated tracheomalacia, it is ideally placed distal to the caudal end of the cleft to minimize contact of the tracheostomy tube with the surgical site. For severe Type IV LTECs, mainstem bronchus intubation may be emergently required to secure the airway.
Medical Management
Type I and some Type II LCs are often approached first with medical management, frequently initiated by a primary care physician, gastroenterologist, or pulmonologist prior to otolaryngology referral for endoscopic evaluation. Control of gastroesophageal reflux is the cornerstone of medical therapy. Proton-pump inhibitors, thickened feeds, upright positioning during feeds, and a speech and swallow evaluation may all be required for optimal reflux control, although it is not uncommon for patients with severe clefts to require ultimately gastrostomy tubes with or without Nissen fundoplication.
Medical management alone may be sufficient for Type I and some Type II LCs. Furthermore, aggressive control of reflux has the added benefit of minimizing inflammatory changes to the cleft tissue and thus optimizing outcomes of surgery if required, as reflux is a risk factor for failure of cleft repair [25]. In patients whose symptoms persist despite medical therapy, early surgery has long been advocated [10]. Controversy remains regarding the regimen and time course of maximal medical therapy, and data conflict as to how many of such patients will ultimately require surgical intervention [8, 9, 21, 22].
Endoscopic Repair
With advances in endoscopic techniques in recent years, the endoscopic surgical approach has been widely applied to Type I LCs, Type II LCs, and some Type III LTECs. The endoscopic approach has several advantages: avoidance of an anterior cervical incision; reduced risk of laryngeal destabilization from an anterior laryngofissure in the setting of a known posterior cricoid deficiency; reduced need for postoperative intubation or tracheotomy; reduced risk of recurrent laryngeal nerve injury; and reduced risk of wound infection, breakdown, or fistula [7].
Prior to endoscopic or open surgery for laryngeal clefts, any other major congenital anomalies that might complicate surgery or anesthesia, such as cardiothoracic pathology, should be addressed first if possible. Laryngeal cleft surgery is ideally performed under general anesthesia with spontaneous ventilation. Inhalation anesthetic agent with judicious oxygen supplementation can be administered via an endotracheal tube placed in the hypopharyx above the larynx. The patient can then be intubated readily for desaturation or decompensation. Alternatively, the use of total intravenous anesthesia without an inhalation agent has also been described, with jet ventilation used intermittently when necessary [26]. The procedure can be performed with the patient intubated when necessary, but exposure is limited and the endotracheal tube can lead to excessive pressure on the surgical wound. In these settings, the smallest endotracheal tube to provide adequate ventilation should be used. The largest laryngoscope that can be accommodated is suspended to maximize surgical exposure.
Since the first report of laryngeal cleft surgery in 1955, the two-layer closure has been advocated and remains the fundamental principle of endoscopic or open surgical repair [27]. Using carbon dioxide laser or cold steel microlaryngeal instruments, two distinct mucosal planes are dissected into the cleft margin: an anterior laryngotracheal mucosal plane and a posterior pharyngoesophageal mucosal plane. Interrupted absorbable sutures are then placed in a caudal to cranial direction, usually posteriorly and then anteriorly for maximal access. Various methods of suturing and knot-tying have been described, and satisfactory results can be achieved using simple or mattress sutures, with either buried or extramucosal knots. Additionally, for Type I LCs, success has also been reported with endoscopic injection laryngoplasty with absorbable gelatin or calcium hydroxylapatite [23].
Open Repair
Open surgery is recommended for some Type III LTECs, Type IV LTECs, and some revision cleft surgery [28]. As these patients may have severe airway compromise, once the airway is secure the procedure may begin with a tracheostomy. Other practitioners have advocated using extracorporeal membrane oxygenation or intraoperative cardiopulmonary bypass to avoid the risks of the endotracheal or tracheostomy tube against the surgical site [17, 29]. Several surgical approaches to the cleft have been described, including an anterior cervical approach with laryngotracheal fissure, an anterior cervicothoracic approach, and a lateral pharyngotomy. The infant trachea is approximately 5 cm from subglottis to carina; therefore, even some Type IV LTECs with low intrathoracic extension can at times be repaired with an anterior cervical approach and no sternotomy or thoracotomy [17, 30]. A novel anterior cervical approach to the Type IV LTEC has recently been described, whereby a cricotracheal separation is performed to reflect the trachea anteriorly and expose the cleft [31].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


