An Overview of Albinism and Its Visual System Manifestations
Elias I. Traboulsi MD
W. Richard Green MD
INTRODUCTION
The term “albinism” (from albus, white) is applied to a group of inherited disorders that are characterized by decreased or absent melanin pigment in tissues, together with developmental abnormalities of the eye and visual pathways. The skin and hair color of patients with albinism depends at least in part on the amount of melanin pigment present and the underlying metabolic defect. Pigmentation can be completely absent throughout the lifetime of the individual, or may increase to nearly normal levels in the first few years of life. In addition to decreased melanin in ocular tissues, patients with albinism have characteristic anatomical defects in the visual system, such as foveal hypoplasia and abnormal decussation of optic nerve fibers. Other common ocular abnormalities include errors of refraction, reduced visual acuity, iris transillumination defects, nystagmus, and strabismus.1,2 The variability in the extent of skin, hair, and eye hypopigmentation, reduction of visual acuity, and nystagmus occasionally makes the diagnosis of albinism challenging. However, a history of marked skin hypopigmentation in infancy, and careful examination for iris transillumination defects, underdevelopment of the fovea, and other subtle clinical signs make it possible to detect mild forms of albinism. Electrophysiologic and molecular genetic testing can be used to confirm a clinical diagnosis in the rare case in which the clinical diagnosis remains in doubt.
A number of inherited systemic disorders feature skin, hair, and ocular hypopigmentation in addition to other abnormalities. Some of these disorders, such as Hermansky-Pudlak’s syndrome (HPS) and Chédiak-Higashi’s syndrome (CHS), are associated with the characteristic ocular and visual system abnormalities of albinism, but others, such as Waardenburg’s syndrome, are not. The latter should not be classified under the general rubric of albinism, but rather as disorders associated with hypopigmentation.3,4,5
FAMOUS ALBINOS
Evidence that Noah (of the Old Testament) was an albino was presented by Sorsby.6 Another famous albino was W.A. Spooner, a British classicist and warden of New College at Oxford University, whose amusing errors of speech came to be known as “spoonerisms.” These aberrations of speech were said to be related to his nystagmus, which caused a jumbling of information from the printed page. Other famous albinos include Edward the Confessor, the Saxon king from 1042 to 1066, and Tamerlane, the central Asian conqueror in the Middle Ages.
EPIDEMIOLOGY
The prevalence of all types of albinism in Northern Ireland has been estimated at 1:10,000.7 In the United States, albinism affects 1:18,000 and 1:10,000 of the white and black populations, respectively. Tyrosinase-positive oculocutaneous albinism (OCA2) is the most common form, with an incidence of 1:37,000 in U.S. whites and 1:15,000 in U.S. blacks. Tyrosinase-negative OCA (OCA1) is the second most common type, and occurs in 1:39,000 U.S. whites and 1:28,000 U.S. blacks. OCA1B (yellow type) is most prevalent among the Amish,8 but has also been reported in other ethnic groups. OCA4 (brown albinism) is more frequent in Nigerians, with an incidence of 1:1,100.9 X-linked Nettleship-Falls ocular albinism (OA) has an estimated incidence of 1:150,000 persons.10 HPS is commonly encountered in Puerto Ricans (1:1,800).11 CHS is very rare, and has been reported in Venezuelan, Caucasian, Japanese, and black patients.12
BIOCHEMISTRY OF MELANIN FORMATION
Melanin synthesis (Fig. 1) starts with the amino acid tyrosine, which is oxidized to dihydroxyphenylalanine (DOPA) by the enzyme tyrosinase. DOPA is then oxidized to dopaquinone by the same enzyme. Dopaquinone is spontaneously transformed to leucodopachrome, and then to dopachrome. The latter is decarboxylated to 5,6-dihydroxyindole, and then oxidized to indole 5,6-quinone. Indole-quinone is converted to melachrome, and then polymerizes to eumelanin (pigment with brown to black color). Dopaquinone can react with cysteine under high concentrations of the latter to form cysteinyl-dopa, which is oxidized to pheomelanin (a pigment with a yellow to red color). DOPA can also react with glutathione to form pheomelanin.
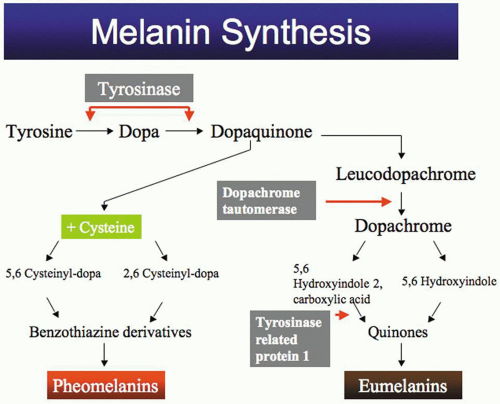 Fig. 1. Melanin synthesis pathway. |
Melanogenesis is identical in melanocytes of the hair, skin, and eye. Melanosome formation begins with the fusion of vesicles from the endoplasmic reticulum and Golgi apparatus. Stage I vesicles contain the enzyme tyrosinase, but no melanin is present. These vesicles fill up with a collection of parallel filamentary structures prior to melanin synthesis (stage II). Melanin granules then deposit on the filaments (immature melanosome, stage III) and progressively saturate them, forming a dense pigmented structure (mature melanosome, stage IV). Skin and hair melanocytes are capable of exporting stage IV melanosomes into adjacent cells, while ocular melanosomes remain within the cell membranes of melanocytes. Melanogenesis in retinal pigment epithelial (RPE) cells is completed shortly after birth, but it may continue for several years in the uveal tract. Patients with OCA have a normal number of melanosomes that are incompletely melanized.
GENETICS OF DISORDERS OF MELANOGENESIS
In 1904 Durham13 discovered that albino animals lacked tyrosinase activity. Kugelman and Van Scott14 later demonstrated, with the use of the hair bulb tyrosine assay, that some albinos showed evidence of tyrosinase activity. This led to the classification of OCA into tyrosinase-negative and tyrosinase-positive categories.
Traditionally, clinicians have divided albino patients into tyrosinase-negative and -positive categories on the basis of clinical findings and the occasional use of the hair-bulb incubation test. They also distinguish between patients with associated systemic disorders (such as HPS or CHS) and those without. These are practical classification schemes, but it would be better to integrate them into one that incorporates recent information on the complex molecular genetics of these disorders. Patients with no tyrosinase activity at all have white hair and no ocular or skin pigment. They have been designated as tyrosinase-negative, while those with any amount of pigmentation have been classified as tyrosinase-positive. The limitation of such a classification is that it relies on clinical features rather than the underlying genetic basis. For example, although the molecular defect in OCA1A and OCA1B is in the enzyme tyrosinase, patients with the former are clinically classified as tyrosinase-negative, and patients with the latter as tyrosinase-positive. So-called autosomal-recessive OA is merely a very mild form of OCA1 in which hypopigmentation is most prominent in the eye, with almost normal skin pigmentation. If strict clinical definition criteria were used, all albinism would be oculocutaneous, except for X-linked Nettleship-Falls OA.
Several genetic loci for human albinism and a spectrum of mutations in each of the responsible genes have been identified in the last two decades (Table 1).5 We now recognize that classic tyrosinase-negative albinism results from mutations that render tyrosinase totally inactive. However, there is a broad spectrum of cutaneous and ocular pigmentation associated with tyrosinase mutations. So-called tyrosinase-positive OCA, in which hair bulb cells form melanin when they are incubated in DOPA or tyrosine, can be caused by mutations in several genes, including tyrosinase, the P gene (vide infra), and others.
Table 1. Molecular Genetic Classification of Nonsyndromic Albinism | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TYROSINASE
The human tyrosinase gene is located on chromosome 11q14-21 and contains 5 exons encoding a protein of 529 amino acids. The tyrosinase polypeptide contains two copper-binding sites, a signal peptide, a region of conserved cysteine residues that create an epidermal growth factor (EGF)-like domain, and a transmembrane region. It is a monophenol mono-oxygenase. A large number of mutations, deletions, and polymorphisms have been discovered in the tyrosinase gene (http://www.cbc.umn.edu/tad/), and recent investigations have tried to elucidate the mechanism by which changes in the tyrosinase protein lead to the clinical manifestations of the disease.15 The majority of tyrosinase gene mutations identified to date have been found in individuals who have the classic tyrosinase-negative phenotype. One type of OCA1—yellow albinism or OCA1B—has been shown to result from a number of tyrosinase mutations that encode an enzyme with less than 10% of residual activity.16 Another OCA1 phenotype results from an R402Q mutation that produces a temperature-sensitive tyrosinase that is active at temperatures below 35°C and is relatively inactive at 37°C.17,18 This is the result of the inefficient exiting of folded tyrosinase from the endoplasmic reticulum at the higher temperature. Individuals with this form of the enzyme have white axillary hair, nearly white scalp hair, light brown arm hair, and dark brown leg hair, and their skin is hypopigmented. Hair color in this variant depends on regional skin temperature. Equivalent animal models include the Siamese cat and the Himalayan mouse.
P GENE
Mutations in the P gene, the human homologue of the mouse pink-eyed dilution gene (p) are responsible for OCA2.19 The human gene is on chromosome 15q and encodes an integral, melanosomal membrane protein of 110 kDa. This protein has 12 putative membrane spanning regions and appears to modulate the processing and transport of tyrosinase.20 The hypopigmentation pattern found in Africans and African-Americans (in whom this is the most common cause of albinism) is characterized by yellow hair and white skin with localized pigmented regions. The irides are partially or completely pigmented with a tan-colored melanin. A 2.7 kb deletion mutation of the P gene is found in 25% to 50% of all African-Americans, and about 80% of South African, Tanzanian, and other sub-Saharan African blacks with OCA2. Mutations in the P gene also cause Brown OCA.21
TYRP1
Rufous OCA occurs in blacks and is characterized by bright copper-red coloration of the skin and hair, and dilution of the color of the iris. It is caused by mutations in the TYRP1 gene.22 TYRP1 has multiple functions, including stabilizing tyrosinase and regulating the production of black eumelanin (see Fig. 1).23
MATP
Mutations in the MATP gene underlie OCA4, a newly identified form of human albinism. OCA1–3 result from the aberrant processing and/or sorting of tyrosinase. The disruption of tyrosinase trafficking occurs at the level of the endoplasmic reticulum (ER) in OCA1 and OCA3, and at the post-Golgi level in OCA2. In OCA4 melanocytes tyrosinase is abnormally secreted from the cells. This process is similar to that seen in OCA2 melanocytes, which results from a mutation of the pink-eyed dilution (p) gene. The P protein and MATP have 12 transmembrane regions and are thought to function as transporters. The abnormal transportation of tyrosinase in OCA4 disrupts the normal maturation process of the melanosomes.24,25 Rundshagen et al26 found mutations in MATP in five of 176 German albinos.
OA1
The gene for X-linked ocular albinism, OA1, was mapped to Xp22.3-p22.2 and was later identified as a membrane protein that is necessary for the maturation of melanosomes.27 In the presence of abnormalities in OA1, melanosomes may fail to bud off the endoplasmic reticulum and enlarge as melanin is deposited, leading to the formation of giant melanosomes (Fig. 2). About 48% of the reported mutations in the OA1 gene are intragenic deletions, and about 43% are point mutations.28,29
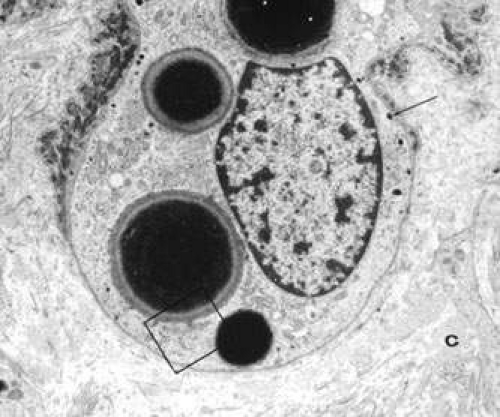 Fig. 2. Electron micrograph of an epidermal melanocyte in Nettleship-Falls X-linked OA. Four macromelanosomes and some normal-sized melanosomes (arrow) are shown (original magnification, 8,000×). (Reprinted from Ref.54.) |
CLINICAL ASPECTS OF OA AND OCA
The severity of the pigmentary dilution in OCA depends on the genetic subtype, constitutive (racial) pigmentation, and age of the patient. Theoretically, a black adult with some tyrosinase acitivity is expected to be darker than a white infant with the same degree of tyrosinase activity. Thus, hair color in OCA ranges from white to dark yellow or light reddish brown (Fig. 3), and iris color ranges from very light blue, with a pink reflex through an undilated iris, to light hazel. The skin may be without pigment or it may have some pigment, especially in nevi and freckles. van Dorp30 observed that some patients with OCA may have normal pigmentation, patients with X-linked OA may be underpigmented, patients with HPS may have a dark complexion, and pigment dilution varies in all types other than OCA1A.
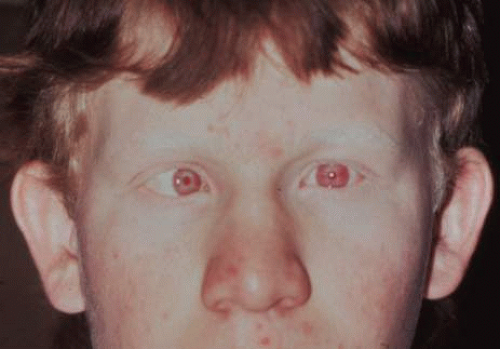 Fig. 3. Teenager with tyrosinase-positive OCA. Note the iris transillumination, esotropia, and reddish-brown hair color. |
The ocular manifestations of patients with albinism vary in severity among the different types of this heterogeneous group of conditions but share characteristic common features.31 Sensory nystagmus is present very early in life but improves in amplitude and frequency with increasing age. This is frequently the presenting sign of the disorder. In most cases the nystagmus is pendular and horizontal. Occasionally a null region and compensatory face turn may be present. A detailed discussion of eye-movement abnormalities in patients with albinism is beyond the scope of this review. Reduced visual acuity is present from birth, and ranges from 20/40 to 20/400 in most cases. Vision usually remains stable, but may improve slightly with age in some patients. Poor binocular vision and reduced stereopsis may be present; however, many patients can pass parts of the clinical stereo tests.32 Iris color ranges from light blue with a pink reflex in OCA1A to hazel in other types, and may change as more pigment is deposited with age. Iris transillumination is best detected by means of the slit-lamp and retroillumination in undilated patients. In some cases the outline of the lens can be seen through the iris (Fig. 4). Hypopigmentation of the iris pigment epithelium results in light scatter and contributes to the subjective complaint of photophobia in some (but certainly not all) patients. Fundus hypopigmentation is also variable. A dilated-fundus examination may reveal a prominent choroidal vascular pattern resulting from decreased or absent RPE melanin (Fig. 5). The optic nerve head may be grayish in patients with OCA1A, or it may be hypoplastic (Fig. 6). Absent macular xanthophyll pigment and a diminished foveal reflex indicate foveal hypoplasia (Fig. 7).33,34 The latter is considered one of the main factors underlying reduced visual acuity in albinism. Abnormal macular vascular patterns may be present in which retinal vessels course through the foveal avascular zone instead of arching around it, which is a hallmark of foveal hypoplasia.35 Whether foveal hypoplasia in albinos is directly related to reduced macular RPE melanin is unknown. Optic nerve hypoplasia is a well documented but not very common finding in albinos.36 Megalocornea, aniridia, posterior embryotoxon, Axenfeld’s anomaly, glaucoma,37,38 retinal coloboma, and abortive cryptophthalmos have also been reported in rare individual patients, and except for mild anterior segment dysgenesis are probably coincidental.
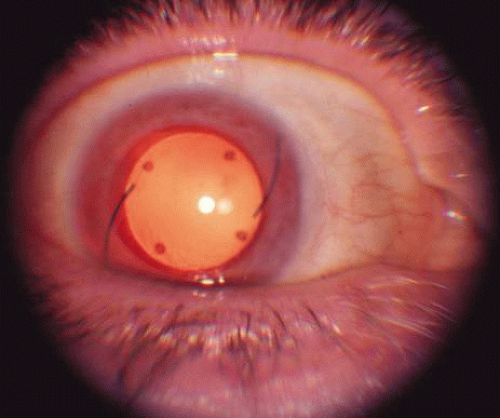 Fig. 4. Retroillumination of the iris in an older patient with OA who had undergone cataract extraction and intraocular lens implantation. The outline of the lens is clearly visible. |
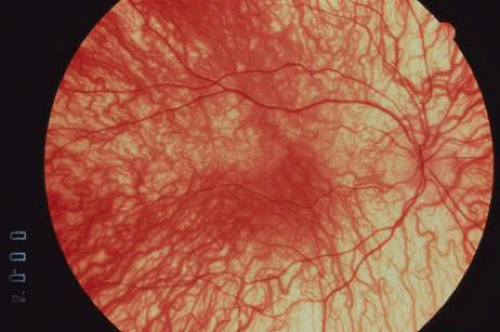 Fig. 5. Prominent choroidal vascular pattern in a patient with OCA. Note the absence of RPE and choroidal pigmentation. |
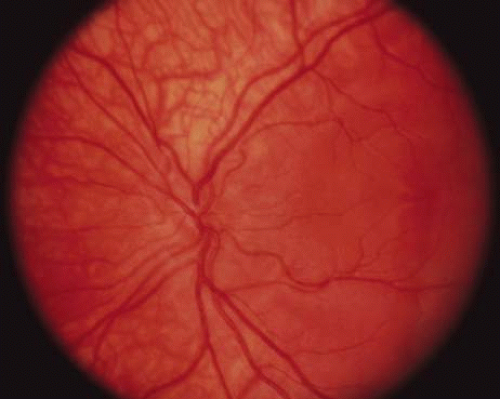 Fig. 6. Optic nerve hypoplasia in a patient with OCA. |
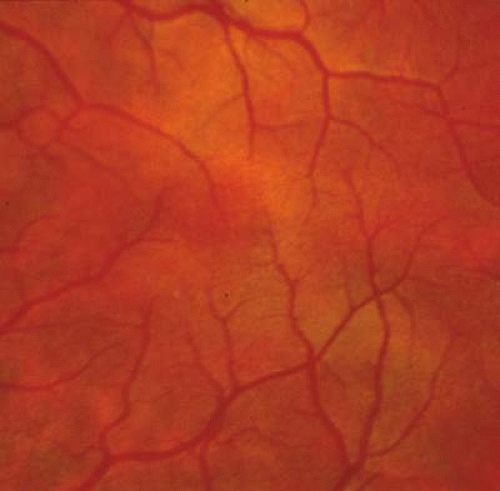 Fig. 7. Foveal hypoplasia in a patient with OCA2. There is no foveal reflex, and some fine blood vessels are crossing the horizontal raphe. |
PATHOPHYSIOLOGY OF VISUAL IMPAIRMENT IN ALBINISM
All albino mammals have abnormalities of visual system development. Three main cellular disorders that lead to reduced vision have been identified: a reduction in the number of rod photoreceptors, the underdevelopment of the central retinal specialization or foveal hypoplasia, and a misrouting of some temporal retinal ganglion cell axons.39 Ganglion cells are produced early in retinal development, and the organization of the retinogeniculate pathway depends on some retinal ganglion cell axons crossing at the optic chiasm to project to the contralateral dorsal lateral geniculate nucleus in the thalamus. In albinos, fewer retinal ganglion cells project to the ipsilateral side, leading to a disruption of lateral geniculate organization and to disorganization of visual information in the cortex (Fig. 8). Tyrosinase activity is required during some phases of the ipsilateral retinogeniculate pathway development. In the RPE, tyrosinase is first expressed on embryonic day 10,40 and pigment formation starts on embryonic day 11.41 The onset of melanin formation starts in the periphery and is graded across the retina. Because melanization occurs at the same time as neuroblast divisions and patterning, tyrosinase expression and the graded onset of pigment formation may be a developmental signal that sets up positional information in the retina. Ganglion cells that project ipsilaterally are produced between embryonic days 11 and 16.41 Tyrosinase expression may also be necessary to ensure that the neuroblasts divide properly. Finally, tyrosinase may be necessary for the pathfinding of the ganglion cell axonal to the lateral geniculate body, which takes place between embryonic days 12.5 and 18.5. After the ganglion cell axons reach the lateral geniculate body, there is a period of refinement of connections and ganglion cell death. Tyrosinase expression in the eye may be necessary to ensure that cells die or are maintained appropriately during this period.42
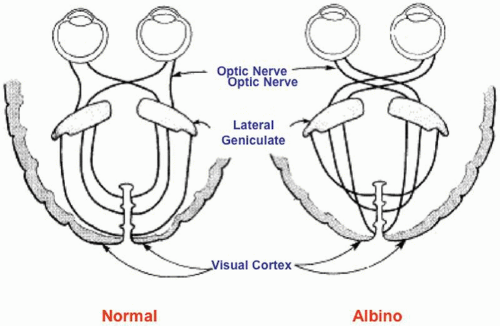 Fig. 8. Pattern of optic nerve fiber decussation in normal (left) and albino (right) individuals. |
ABNORMAL DECUSSATION OF OPTIC NERVE FIBERS
Carroll and colleagues43 presented evidence that human albinos have the same misrouting of the retinogeniculate projections that has been found in albinos of other species. When pigmentation is incomplete, the developing optic tracts revert to an almost completely crossed pattern at the chiasm, as is the case in more primitive panoramic systems. In normal humans, 45% of the axons that originate in the temporal half of the retina remain uncrossed as they pass through the chiasm and project to the ipsilateral lateral geniculate nucleus. The greatest number of these fibers subserves the central 20° of the temporal retina. In albinos, most of these fibers decussate at the chiasm and synapse in the contralateral lateral geniculate nucleus, leaving only 10% to 20% of fibers uncrossed (Fig. 8). These abnormalities of decussation lead to a predominantly monocular representation of the central visual field in each occipital cortex. Monocular visual-evoked responses hence lead to markedly asymmetric responses from the two hemispheres.44,45 All OCA or OA conditions in humans and animals with nystagmus tested to date have shown either electrophysiologic or anatomic evidence of a decussation defect in the optic tracts. Evidence that anomalous decussation also exists in the auditory system was presented by Creel and colleagues.46 It is thought that 1% to 2% of the human population may be heterozygous for albinism, and that this may have an adverse effect on binocular depth perception. The locus ceruleus and substantia nigra are normally pigmented in albinos. They owe their pigmentation to neuromelanin, which is synthesized by tyrosine hydroxylase rather than tyrosinase.
LIGHT SCATTERING
Light scattering within the eye may play some role in the pathophysiology of visual impairment in albinism, but its contribution must be minimal. For example, patients with autosomal-dominant OCA have translucent irides and an albinotic fundus, but normal visual acuity. Some of these patients complain of photophobia, which may be a consequence of light scattering. Tinted lenses can help. Corneal contact lenses with an opaque iris portion have not significantly improved visual acuity in albinos.47
LIGHT-INDUCED RETINAL DAMAGE
Light-induced retinal damage does not appear to play any appreciable role in the visual impairment of albinos. There is histopathologic and ultrastructural evidence that extraordinarily intense light can produce nonthermal retinal damage even in normally pigmented animals.48 Albino animals have lower thresholds for such damage. This has been attributed to an ineffectual iris diaphragm, because normally pigmented animals show somewhat compatible thresholds with pupillary dilatation. There are no data to suggest that visual function in human albinos deteriorates with age. Nevertheless, it seems reasonable to limit exposure to unnatural, moderately intense light sources. It has been hypothesized that light-generated free radicals are responsible for nonthermal light damage. Feeney and Berman49 suggested that melanin normally plays a protective role by reducing such free radicals.49
CONGENITAL NYSTAGMUS
The nystagmus that is invariably present from birth in albinism may contribute to visual impairment, and is occasionally misdiagnosed as congenital motor nystagmus or nystagmus secondary to retinal dystrophy. Black patients with OA are often misdiagnosed as having motor-defect congenital nystagmus. Contrary to the clinical impression, waveform analysis does not confirm a consistent difference in the type of nystagmus between sensorial causes of congenital nystagmus (such as albinism) and the primary motor defect type of congenital nystagmus.50 Both pendular and jerk-type patterns can be found in each group, depending on the test conditions. Also, in both groups the nystagmus consists of horizontal and rotary excursions. In motor-defect congenital nystagmus, some patients assume a compensatory head position to find a zone of least nystagmus and best visual acuity. Eye muscle surgery on such patients to shift the null point to the primary position of gaze can be effective in eliminating abnormal head positions and improving function. Although albinos typically do not assume such head positions, the author has observed a number of such patients. An occasional patient with albinism may require eye muscle surgery to correct the torticollis. An improvement in visual acuity is not to be expected, because of the sensorial defect. Four-rectus-muscle maximal recession, and disinsertion and reattachment of the rectus muscles to their original insertion site have been advocated as effective methods to reduce the amplitude of nystagmus and improve the vision of patients with albinism. The experience with such procedures to date has been very limited, and they have not been widely adopted.
MACULAR HYPOPLASIA
Macular hypoplasia is probably the most significant vision-limiting factor in albinos. The clinical presence of presumed “hypoplasia” has been appreciated for a long time, but it was difficult to interpret early histopathologic studies of albino eyes. There seems to be a consensus that the macular luteal pigment is absent. Postmortem serial sections through the presumed macular retina of the eyes of oculocutaneous albinos51,52,53 (Fig. 9) and an ocular albino54 have been studied. In each case, no foveal pit or umbo was found, but the central ganglion and nuclear cell layers were present and of normal thickness. Mietz and co-workers53 examined the eyes of a 99-year-old tyrosinase-negative albino woman. They found a posterior embryotoxon, absence of melanin in all ocular structures, and absence of a foveal pit. In the normal anatomic position of the foveola, the retina was of normal thickness, with five to seven ganglion cell layers. In another study, the morphology of the outer segments in the presumed foveal area suggested an absence of the rod-free area, and the presumed foveal cones resembled extrafoveal cones in shape.52 Optical coherence tomography has also shown the absence of a foveal pit in albinism.55 The etiology of foveal hypoplasia in albinism is thought to be related to the decreased amount of melanin in the retinal pigment epithelium.
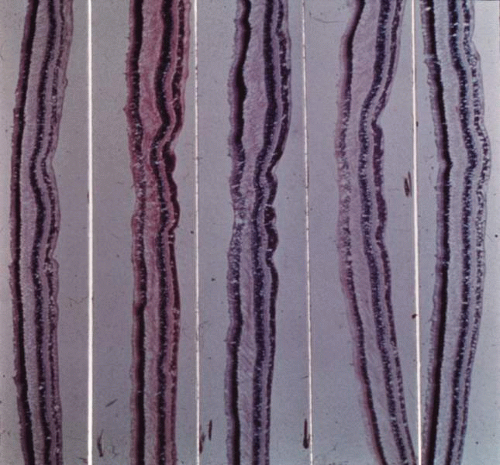 Fig. 9. Sections through the center of the anatomic location of the fovea reveal no foveal pit. (Reprinted from Ref.51.) |
In the following section the salient clinical features of the various types of albinism are discussed.
TYROSINASE-POSITIVE OCA (OCA2)
OCA2 is the most common form of albinism. It is clinically differentiated from tyrosinase-negative albinism or OCA1A by the presence of some pigment in the hair, skin, and eyes of all patients beyond infancy (Fig. 3). Pigmented nevi and freckles develop around the age of 5 to 6 years. There is susceptibility to basal cell and squamous cell carcinoma of the skin, and cutaneous malignant melanoma has been reported in a few cases.56,57 Electron microscopy reveals lightly melanized melanosomes in the skin and hair of older patients. Infants with OCA2 may be clinically indistinguishable from those with OCA1A, but they acquire pigmentation with age. The hair bulb incubation test or mutation analysis of the tyrosinase and P genes can help differentiate the two disorders in infancy. Mutations in the P gene in patients with tyrosinase-positive OCA suggest that at least in some patients, albinism is due to a decreased availability of tyrosine in the melanocytes because of a problem in its transportation into the cell.58,59 The P protein is thought to be important in the transport of tyrosine. In the mouse, the p (pink-eyed dilution) mutation results in albinism. OCA2 maps to 15q11.2-q12, a region associated with Prader-Willi’s and Angelman’s syndromes, both of which are characterized by hypopigmentation in addition to dysmorphic and developmental abnormalities.60,61
TYROSINASE-NEGATIVE OCA (OCA1)
OCA1 is the second most common type of OCA. Patients with OCA1 do not have clinically or histopathologically discernible pigmentation. Pigmented nevi and freckles do not occur. Electron microscopy of the skin and hair reveals no melanization of melanosomes. In vitro hair bulb incubation in L-tyrosine does not produce melanin. There is a total absence of tyrosinase enzyme activity. In a study of tyrosinase gene mutations, Tripathi and co-workers62 concluded that in Caucasians OCA1 results from a great variety of uncommon alleles. About 90% of the patients in their study had a total of 29 mutations, and 80% of missense substitutions were clustered in two relatively small regions of the tyrosinase polypeptide. Not surprisingly, in Tripathi et al’s study, six of 17 patients who were initially classified as having tyrosinase-positive OCA had mutations in the tyrosinase gene. Several other review studies of mutations in the tyrosinase gene have been published.63,64,65,66
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


