10 Acromegaly
Although acromegaly has been recognized as an entity for a very long time, it was first clinically described by Pierre Marie1 in 1886. Initially acromegaly was thought to be a disease, primarily bone hypertrophy, and pituitary gland enlargement was regarded as a further manifestation of generalized bodily organ enlargement.2,3 Massalongo4 was the first to suggest that the pituitary gland may be the cause of the disease, and in 1900 Benda5 postulated that the eosinic granules that were observed in the glands of acromegalics were secretory. In the same year that Massalongo first made his assertion, F. T. Paul attempted a temporal craniotomy to remove a large pituitary tumor. Although he was unsuccessful at resecting the tumor, the patient apparently did benefit from the subtemporal decompression.6 Subsequent to this success there was an increase in transcranial approaches to the gland, but surgery was usually reserved for treatment of mass effect. In 1908 Hochenegg7 performed the first transsphenoidal operation for acromegaly, and one year later Harvey Cushing8 did his first transsphenoidal operation for acromegaly in the United States. The transsphenoidal approach, although initially well accepted, fell out of favor in exchange for the transcranial approaches, and due to the rather high morbidity and relatively poor results, surgical treatment of acromegaly was employed only for decompression for mass effect.9 Most patients were treated with radiation or medical therapy. In 1963 the first growth hormone (GH) assay was developed, and it enabled the clinician to assess the treatment of the disease.10,11 As it became apparent that the available treatments at the time were far from optimal, the time was right for the reintroduction of the transsphenoidal approach for the treatment of acromegaly. Gerard Guiout12–14 and his pupil Jules Hardy,15–17 incorporating the operative microscope and fluoroscopy, were the first to demonstrate the safety and efficacy of the procedure for selective resection of pituitary tumors while preserving normal pituitary gland function. Over the past several decades the techniques have been refined and the procedure has reemerged as the firstline treatment to cure GH-secreting adenomas.18,19
Patients with acromegaly are at significantly increased risk of morbidity and mortality compared with an age-matched population. The deleterious effects of chronic GH and its downstream hormone insulin-like growth factor-1 (IGF-1), formerly known as somatomedin C, cause damage to all of the organs in the body, and the increased risk of death is largely due to an increase in cardiovascular disease in the acromegalic population.
 Presentation
Presentation
Acromegaly is found in the general population at a rate of 50 to 70 cases per million with an incidence of 3 to 4 cases per million per year.20–22 Patients typically present with the disease between ages 40 and 50. GH-secreting pituitary adenomas make up approximately 16% of surgical cases of pituitary lesions.23 Acromegalic patients have between a 1.9- and 3.3-fold increased mortality rate.
Acromegaly is a disease of chronic overproduction of GH. In the child and preadolescent, excess GH leads to abnormally increased height and gigantism as well as organomegaly. Once the epiphyseal plates have closed, the manifestations are that of acromegaly. The consequences of GH hypersecretion are numerous. Bone and soft tissue overgrowth is almost always present. Patients may first notice changes in shoe size, ring size, and a coarsening of their facial features, with enlargement of their lips, macroglossia, a prominent brow, prognathism, and increased spaces between their teeth. A low voice is a product of laryngeal hypertrophy. Patients go on to develop skeletal abnormalities secondary to new bone growth, including vertebral body osteophyte formation leading to spinal stenosis, and bowing of the long bones. Overgrowth contributes to a significant increase in weight and stress on the joints, cartilaginous erosion, and severe osteoarthritis. Acromegaly causes numerous cardiovascular complications, such as cardiomyopathy, arrhythmias, and coronary artery disease.24–26 Patients with acromegaly develop impaired diastolic cardiac function even in the absence of frank cardiomyopathy.25 GH elevation contributes to markedly increased rates of diabetes and hypertension. Interestingly, patients with acromegalyless commonly present with significant panhypopituitarism than do those with nonsecreting tumors.27 Although there is an increased frequency of hypothyroidism, patients with acromegaly are less likely to have hypothyroidism, impaired adrenal function, or hypogonadal function than similar patients with nonsecreting pituitary adenomas. This remains true even when macroadenomas are independently compared in both groups. This may be due to secondary effects of GH. IGF-1 has been observed in vitro to stimulate DNA28,29 and thyroglobulin synthesis.30
The vast majority of patients presenting with acromegaly have an underlying pituitary tumor. Rarely the disease can be caused by growth hormone—releasing hormone (GHRH) elevation secondary to hypothalamic dysfunction.31 Very rarely the disease can also be caused by ectopic release of GHRH by carcinoid or pancreatic islet tumors.32–34 Although these are truly the exceptions, the possibility must be considered to avoid potentially unnecessary pituitary surgery. Furthermore, glands that have been biopsied under these circumstances demonstrate hyperplasia without evidence of adenomatous changes.34 Clearly, increased stimulation of the gland by GHRH is not enough to produce an adenoma.
Mortality is significantly increased in patients with untreated or uncured acromegaly. Retrospective data from New Zealand showed a significant increase in the death rate from cardiovascular complications. Analysis revealed that there were statistically significant increases in survival with decreasing GH levels. At random GH levels of 1 μg/L, the death rate approaches that of the general population.35 Reduction of IGF-1 to the normal range produced a mortality equivalent to that of the general population. Other studies have found that mortality approximated that of the general population when GH approached 2.5 μg/L36 or 5 μg/L.37 Most recent studies confirm that a lower GH is associated with a better long-term morbidity and a reduced mortality rate. Cardiomyopathy has been shown to reduce significantly when IGF-1 levels are normalized and GH levels are <1 μg/L after an oral glucose load.25 Patients with uncured acromegaly have not only an increased cardiac risks but also an increased risk of respiratory and cerebrovascular death. These increases may not decrease fully with normalization of GH levels.36 This is speculated to be due to the fact that some of the processes associated with acromegaly, such as diabetes and hypertension, may have already caused significant endorgan damage by the time that GH levels are normalized, so that the patient continues to experienced an increased risk of death. The association between acromegaly and malignancy is unclear. Orme et al36 performed a large retrospective review of 1362 patients with acromegaly and found a higher than expected rate of colon cancer in patients with acromegaly but a lower than expected overall incidence of malignancy in this population. Increased colonic adenocarcinoma rates have been found in other series as well.37–39 Other studies have failed to document this increased rate of malignancy.35
Reduction in morbidity and mortality relies on prompt diagnosis and early intervention and treatment.18 Patients frequently present early to their primary care physicians with protean complaints, and it is incumbent upon physicians to recognize the disease in its earliest manifestations and refer the patient for definitive care.
 Diagnosis
Diagnosis
Growth hormone is under the control of the hypothalamic hormone GHRH. The pituitary somatotrophs and adenomas secrete multiple isoforms of GH. The predominant one is a 22-kd monomeric form accounting for approximately 50% of the circulating GH in most patients, but the other monomers are also active. Different assays have different sensitivity and specificity for the different monomers of GH. Because of this there still exists test-to-test variation until adequate standardization is employed.40
Hypersecretion of GH leads to increased production of IGF-1. In the past, diagnosis of acromegaly relied solely on the recognition of the constellation of physical signs and symptoms. In the 1960s the advent of radioimmunoassay allowed for the measurement of GH levels, and following that similar tests were developed to measure IGF-1 levels.41 As these tests have been refined and their sensitivity increased, it has prompted further evaluations of what is considered “normal” levels of these hormones.42 Accurate measurements of these biochemical markers allows for earlier diagnosis of the patient and assessment of cure.
Growth hormone has a half-life of 15 to 20 minutes and is released in a pulsatile fashion secondary to stimulation by GHRH. It is inhibited by the release of somatostatin. Acromegaly can be characterized by increased frequency of GH pulses, increased mean GH nadir, increased nonpulsatile GH fraction, and an increase in the “disorderliness” of GH release.43–45 In the normal individual GH ranges from <0.1 to 0.2 μg/L interspersed with periods with levels as high as 20 to 30 μg/L. Although individuals with exceedingly high random levels of GH may be considered likely to have acromegaly, especially if clinically suspected, there is some limitation to interpreting a random GH sample.41 More effective and popular is the measurement of GH levels after an oral glucose load. In the normal individual GH levels will drop significantly following ingestion of 75 to 100 g of glucose. The criteria for diagnosis of acromegaly have changed over the years. In the 1980s levels of <10 μg/L of GH on random assay were considered within normal limits,46 and <2 μg/L after oral glucose was also considered normal.47 Recent data have set the cutoff for a random level at 2.5 μg/L, and the cutoff for a normal suppression test at 1 μg/L.48
As IGF-1 is the “downstream” hormone of GH, there is reason to think that continuing to measure the IGF-1 levels may provide more reliability in the consideration of remission in the acromegalic. Up to 50% of patients with GH nadirs of <1 μg/L still have elevated IGF-1.42 A subset of patients also exists with the obvious stigmata of acromegaly and IGF-1 elevation but with normal mean GH levels.47 The need to measure IGF-1 levels becomes further necessary as the new competitive GH antagonist pegvisomant has been introduced, which results in elevated GH levels accompanied by a decrease in IGF-1 levels. IGF-1 has a significantly longer half-life, and therefore random measurement is more feasible to assess posttreatment acromegalics. Current recommendations are that IGF-1 levels be at normal age- and sex-adjusted levels to define remission. The patient’s risk of mortality has been shown to approach that of the general population when the IGF-1 level is normalized.49 However, IGF-1 levels do not increase significantly once GH levels are <20 μg/L, and because of this IGF-1 in the absence of GH measurement will not be effective in assessing partial responses to therapy when the GH levels remain high.
Because of the complementary nature of information provided by IGF-1 and GH levels, it seems prudent to continue to measure both sets of data in posttreatment patients to properly assess and manage them, and currently this remains our protocol.
 Histopathology and Prognosis
Histopathology and Prognosis
Microscopically, the appearance of adenoma is very varied. Cells may be round or ovoid. They may be arranged in monotonous sheets or show acinar or papillary patterns. The nuclei may be cytologically pleomorphic with mitotic figures and a stippled chromatin appearance. Invasion of local dura may be demonstrated even in cells with bland cytologic features.50 GH-secreting tumors can be categorized into two distinct morphologic types: densely granulated (DG) and sparsely granulated (SG) adenomas (see Chapter 6). DG cells resemble the well-differentiated somatotroph cells of the normal anterior pituitary gland, whereas in contrast the SG cells bear little resemblance to the normal GH-secreting cells.51 SG cells have cytoplasmic cytokeratin aggregates sometimes called fibrous bodies or intracytoplasmic filamentous aggregates. The two subtypes of tumors have been reported to occur in relatively equal frequency.52 SG adenomas tend to grow more rapidly and are more likely to be invasive at the time of diagnosis.53
Recent studies have suggested that the Ki-67 labeling index may predict clinical outcome in the postsurgical management of acromegalic patients, with a lower index correlating with a higher remission rate.54
 Treatment
Treatment
The goals of treatment are control of GH and IGF-1 levels such that GH levels are <1 μg/L after an oral glucose load and that IGF-1 levels are normal, adjusted for age and sex. The tumor mass needs to be removed or significantly enough reduced so that there is no chance of recurrence and so that the optic nerves are free of compression or impending threat of such.48 It is common that acromegaly will require multimodality therapy.55
Surgery
Transsphenoidal microsurgery is the preferred primary treatment for acromegaly, though craniotomy rarely is indicated as a primary surgical intervention.48,49,56–59 We prefer a transseptal endonasal approach to the sphenoid. Patients with significant bony and cartilaginous hypertrophy with small nares may provide more of a challenge for maneuvering of the instruments, but with a well-placed speculum we usually find that the nasal route still affords adequate surgical exposure of the sella and enough space to safely remove tumor. Nonetheless, we are prepared to convert to a sublabial approach should we find that the exposure is unsatisfactory for tumor resection, though we very rarely need to do this. We make no significant modifications to our standard approach to the sphenoid for acromegalics. We do not employ lumbar drainage as a standard part of our protocol. We believe that adequate exposure of the sella and surrounding structures can be afforded by the microscope or the endoscope, but there are situations where some may find the endoscope beneficial to assess for completeness of tumor resection after microscopic removal.
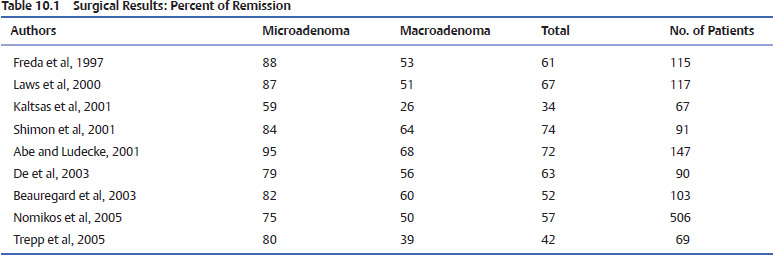
Surgical resection of the adenoma provides the highest hope for cure and is generally safe with acceptable morbidity.49,56–58,60–63 As previously discussed the definition of biochemical cure in the acromegalic patients has been evolving. Older series of transsphenoidal surgery for acromegaly, which incorporated now outdated definitions of cure, may obscure the interpretation of the success of surgery for GHsecreting tumors in the literature. Early studies used GH levels of <5 μg/L or <10 μg/L as the definition of cure58,64 When these numbers were employed, cure rates ranged between 67 and 90%.63,65,66 Rates of cure for acromegaly by transsphenoidal surgery as defined by a GH of <5 μg/L ranged from 5315 to 81%.63 Abosch et al,56 using as a definition of cure a GH of <5.0 μg/L, found a 76% remission rate. Age, tumor size, grade, and preoperative GH levels were all predictive of remission. Of the patients who were defined as being in remission, 13% nonetheless had elevated IGF-1 levels (Table 10.1).
Tumor size obviously influences the likelihood of remission. The odds of remission for microadenomas was found to be 3.3 times higher than the odds for macroadenomas.49 The success rate for modern microadenoma resection alone ranges from 74 to 88%.56,67,68 Success rates for surgery on macroadenomas are, as expected, lower than those for smaller tumors. Macrodenomas that are noninvasive have modestly lower remission rates of 67%,68 whereas those that invade the cavernous sinus and other adjacent structures have rates as low as or lower than 30%. Further analysis revealed the rather unsurprising fact that tumors can further be subclassified beyond micro- and macroadenoma, and that increasing size correlates with decreasing success rate. Major decreases in remission exist when the tumor was invasive and >20 mm. GH rates may be predictive of the likelihood of success. According to Shimon et al,69 although patients with a preoperative GH level of <20 μg/L or of 20 to 50 μg/L had cure rates of 90% or 79%, respectively, those with a GH level of >50 μg/L had a cure rate of only 16%. Similarly, Freda et al68 found 80% and 90% cure rates of micro- and macroadenomas, respectively, when the preoperative GH level was <10 μg/L, falling to 55% for tumors with a GH of 30 to 50 μg/L. No patients were cured with a preoperative GH of >200 μg/L. Because GH levels rapidly fall following resection, whereas IGF-1 can take a long time to normalize, GH levels are routinely the test of choice in the immediate post-operative period. Early GH levels can be predictive of success or failure. There is no clear cutoff on the early postoperative laboratory results to define cure, but GH levels of <2 μg/L on the first postoperative day are found to correlate with surgical success in 99% of patients.67 GH levels of <3 μg/L predict cure in 89%,68 whereas a GH of >5 μg/L in the early postoperative period is associated with much poorer normalization rates of IGF-1 of 0 to 21%.58,68,70 According to Krieger et al,67 only one of 65 patients with a GH level of >2 μg/L on postoperative day one was later determined to have a biochemical cure. Dural invasion and extrasellar extension of tumors have been associated with persistent disease.
Patients with acromegaly usually present for endocrinologic disturbances. Some patients present with visual disturbances. Most patients recover some degree of visual function following trans-sphenoidal surgery. The recovery of visual function relates to the time of optic nerve compression and the ability to adequately decompress the nerves in the operating room. As patients with acromegaly largely have other symptoms as well, they are more likely recognized at an earlier point in their disease. Nonetheless, some patients do present with significant nerve compression or pituitary apoplexy, leading to acute visual deterioration; 80 to 86% of patients presenting for primary surgery for visual disturbance due to various lesions of the sella typically improve postoperatively.71,72 In one study, 75% of patients had an improved visual examination following transsphenoidal surgery for acromegaly.61
Some patients do not present until they are elderly; as many as 5% of patients with acromegaly present after the age of 65.73,74 Many clinicians are tempted to steer these patients away from surgical resection because of the belief that they are unable to handle the stresses of surgery. Puchner et al74 report very good results for patients over the age of 65 undergoing surgery for acromegaly. They report no significant perioperative morbidity and no mortality. Results of resection are extremely good. As patients can experience reversal of the effects of acromegaly within 6 months to 1 year following successful surgery, even elderly patients may benefit. These patients may experience improvement in their cardiac function, glucose tolerance, and hypertension, all of which may improve life expectancy and quality of life. Thus, many elderly patients, despite their acromegaly and its complications, are deemed to be acceptable surgical candidates.75
Reoperation for acromegaly is a controversial subject. Rates of surgical success are poor, probably because many patients have extensive tumor involvement that is not as likely to be amenable to curative resection. Rates of surgical success for endocrine cure range from 0 to 57%.62,69,76 Morbidity associated with reoperation is much higher; 63% of patients develop hypopituitarism.62 One group classified reoperative tumors according to size and invasiveness and documented significant success with the microadenomas and those that radiographically did not appear to be invasive.76 However, the group had no successes reoperating on those tumors already deemed to be invasive. Some patients need to be reoperated on for progressive visual deterioration. Patients undergoing reoperation for impairment do not generally seem to garner as much visual improvement as those undergoing a primary operation with the same complaint.72
Recurrence of active acromegaly is a confusing issue. As testing becomes more sensitive, more patients are noted to have subtle signs of endocrinologic laboratory abnormality. Some patients now considered to have had a recurrence may just have had mild GH dysfunction that was not detected as abnormal by less accurate earlier tests. More contemporary literature has defined the rate of recurrence as between 1.1 and 19%.49,56,61,68,77 Patients who experience a recurrence are typically managed in multimodality therapy with medical and radiation therapy.56 Recently we reported on a subset of patients with normalized IGF-1 levels postoperatively but who failed to suppress to levels of <1 μg/L; five of 14 of these patients later developed evidence of elevated IGF-1.78
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


