Molecular Genetics and Pharmacogenomics of the Glaucomas
This chapter introduces the reader to the shift from a “single gene, rare disease” concept to a “complex and multiple gene disease” model. By reading this chapter, you will learn about the expectations of how genomic testing will pave the way to individualized treatment for patients with various forms of glaucoma. It begins with highlighting the difference between single genes, which when mutated may result in striking clinical phenotypes (e.g., Axenfeld–Rieger syndrome), versus genes that may have DNA sequence variants (known as polymorphisms) that, with or without environmental contributions, can be associated with more common forms of glaucoma (e.g., exfoliation syndrome). Insights into the etiology and pathogenesis of various forms of glaucoma gleaned from analysis of DNA, RNA, or protein are then described. These insights will likely lead to new targets for glaucoma therapy that are beyond simply lowering intraocular pressure (IOP). The chapter ends with a discussion of pharmacogenomics and how genomic testing may help clinicians develop more rational, personalized treatment for their patients.
This chapter begins with three cases to illustrate the promising application of molecular medicine in the clinical context.
CASES
Please review each of these clinical scenarios and keep them in mind as you go through this chapter. Comments will be made on each of these cases later in the chapter.
Case 1
A 17-year-old female patient presents to your office reporting blurred and gradually decreasing vision. On examination, her visual acuity is 20/20 OU and the IOP is 30 mm Hg OU. Her angles are open and normal by gonioscopy. Central corneal thicknesses measure 503 µm OD and 498 µm OS. She has near-total cupping of both optic nerves (Fig. 8.1A). Visual field testing demonstrates defects within 10 degrees of fixation OU (Fig. 8.1B).
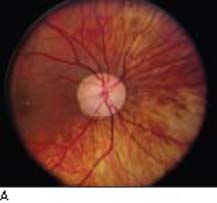
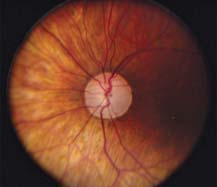
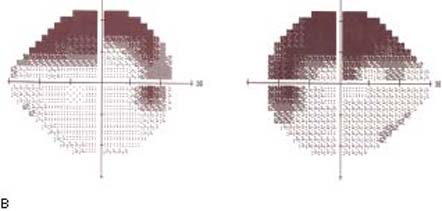
Figure 8.1 Optic disc photos (A) showing very thin neuroretinal rim in each eye. Visual fields (B) showing advanced nerve fiber bundle defects encroaching on fixation in the left visual field and within 10 degrees in the right visual field.
The authors gratefully acknowledge David Murrel and David M. Reed for their assistance with artwork in Chapters 8 and 27
On inquiring further, you learn that her mother and sister also have glaucoma that developed relatively early in life. The mother is blind in one eye, and the sister’s eyes are stable after having glaucoma surgery in both eyes.
The patient asks several insightful questions:
- What do I have?
- Will I go blind if I don’t receive treatment, and what is my best treatment option?
- What are the chances that any future biological children of mine would also get this disease?
- Can anything be done other than medications and surgery to treat my condition?
Case 2
A 40-year-old Scandinavian man has a mother with advanced exfoliative glaucoma (Fig. 8.2). He wants to know his chances of developing the same condition.
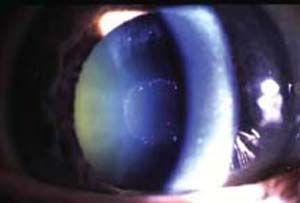
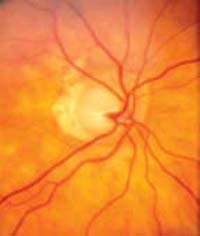
Figure 8.2 Appearance of the anterior and posterior segments of the mother’s right eye.
Case 3
A 68-year-old woman presents for advice about her glaucoma diagnosis and its impact on her children. She brings along her personal “smart card” that contains her medical history, past visual fields, optic disc imaging, and genomic sequence.
At diagnosis, her IOPs measured 33 mm Hg in both eyes, and her central corneal thickness measurements were 584 µm OD and 566 µm OS. She was otherwise asymptomatic, and she was treated for glaucoma on the basis of the appearance of the neuroretinal rim of her optic disc (Fig. 8.3).
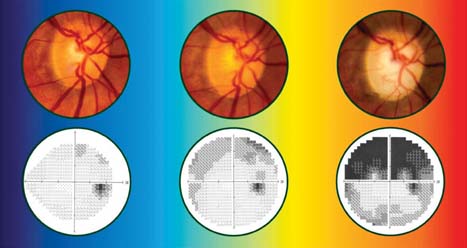
Figure 8.3 Case demonstrating progression of glaucoma based on right optic disc photos (A) and right visual fields (B) over 18 years despite medical and surgical treatments with IOP reduction and fluctuation between 7 and 13 mm Hg. (Modified from Moroi SE, Richards JE. Glaucoma and genomic medicine. Glaucoma Today. 2008;1:16–24, with permission.)
Over the following 18 years, her IOPs fluctuated between 7 mm Hg and 13 mm Hg with medical and surgical treatments. Despite this management, she developed progressive cupping of the optic disc and visual field loss (Fig. 8.3, center and right) over time. She asks: “Will the same thing happen to my children?”
THE HUMAN GENOME
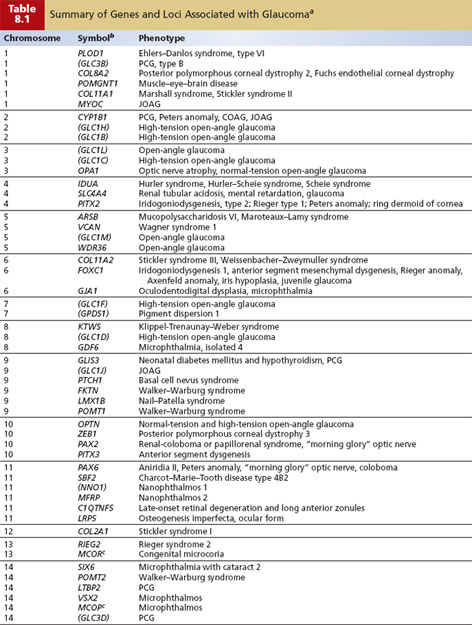
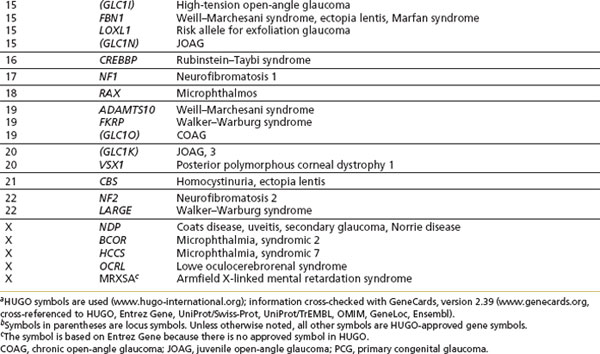
Genes for glaucoma are found throughout the human genome (Fig. 8.4). There are approximately 20,500 genes encoded in the 6 billion base pairs that make up human DNA distributed on 46 chromosomes (1). In addition, 37 “mitochondrial” genes are encoded in the circular mitochondrial DNA that is inherited through the mother. An offshoot of the Human Genome Project (http://www.genome.gov/10001772) was the International HapMap project (http://www.hapmap.org/), which permitted the identification and cataloguing of genetic sequence variants among individuals across diverse populations. These variants are known as single-nucleotide polymorphisms, or SNPs (pronounced “snips”). These SNPs are recognized as markers for chromosomal regions where genetic variants are shared among individuals of a given ethnic group. By taking advantage of these conserved DNA blocks marked by these SNPs, early successes have shown promise to identify certain SNPs as potential markers for disease. Future research may shed further insight on disease onset, disease severity, and treatment response, thus paving the way toward the advent of “personalized medicine.”
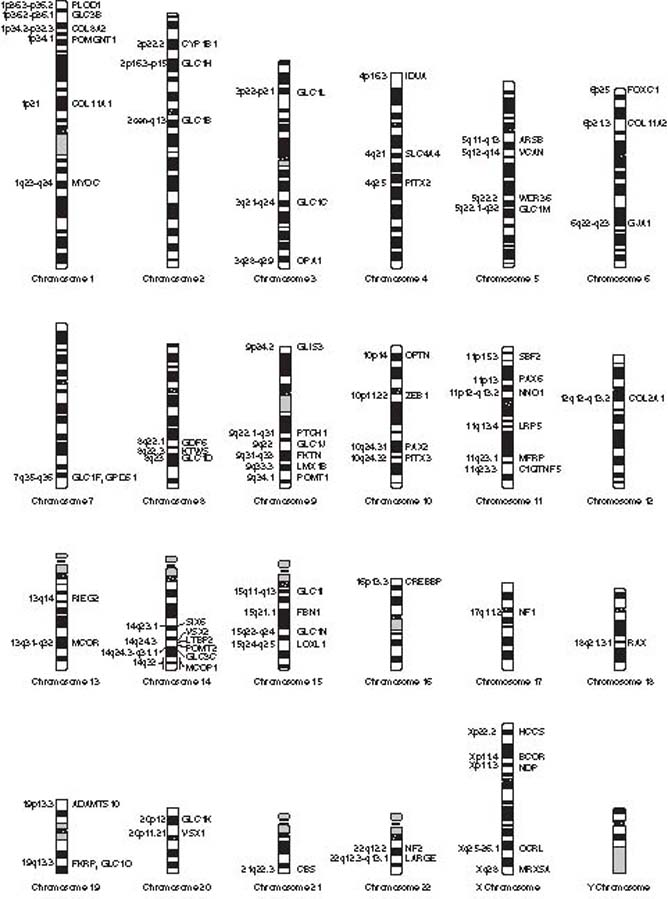
Figure 8.4 Chromosomal location of genes and loci for various forms of open- or closed-angle glaucoma are found throughout the human genome. Only the Y chromosome is believed not to harbor a gene or locus for glaucoma.
Mendelian (“Single Gene”) versus Non-Mendelian (Complex) Diseases
Mendelian disorders are typically rare diseases that follow Mendelian patterns of inheritance—the laws of segregation of alleles and the law of independent assortment. Common examples of Mendelian patterns of inheritance include autosomal-dominant, autosomal-recessive, and X-linked inheritance. Clinicians are familiar with these rare or uncommon clinical disorders because of the striking clinical phenotypes, such as juvenile open-angle glaucoma (JOAG), illustrated in Case 1, and others involving anterior segment dysgenesis, such as Axenfeld–Rieger syndrome. The genetics of such cases represent the “single gene—single disease” model.
In contrast, non-Mendelian, or complex, disorders do not follow the classical rules of Mendelian inheritance. Examples include quantitative traits that result from the additive effects of many genetic or environmental effects, polygenic traits that happen only if defects are present in more than one gene, traits displaying incomplete penetrance, codominant inheritance in which each of the three genotypic combinations for an allele have a different phenotype, imprinting effects caused by chemical modifications to the DNA, or mitochondrial inheritance. Representative conditions and diseases include exfoliation, normal-tension glaucoma, and chronic open-angle glaucoma (COAG).
There are various approaches used to identify a single gene or multiple genes that are involved in inherited disorders. These approaches can also be applied to the discovery of genes underlying treatment outcomes in the field of pharmacogenetics (how an individual’s genes affect the way the individual’s body responds to a medication or treatment) and pharmacogenomics (the study of drug responses in the context of the entire genome). (The topic of pharmacogenetics and pharmacogenomics is addressed later in this chapter.) The selection of a particular approach or method depends on the frequency of the disease mutation and the penetrance of the mutation (the frequency with which the presence of a particular genotype in an organism results in the corresponding phenotype) (Fig. 8.5).
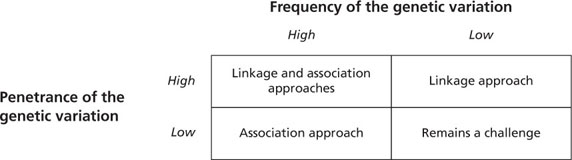
Figure 8.5 Overview of application of linkage and association approaches to identify genes as markers for complex diseases and quantitative traits. The appropriate approach selected for a study depends on the frequency of the genetic variant and the penetrance of the disease mutation. (Modified from Moroi SM, Raoof DA, Reed DM, et al. Progress toward personalized medicine for glaucoma. Expert Rev Ophthalmol. 2009;4(2):145–161.)
Two common approaches used to identify genetic variants that contribute to inherited diseases are termed linkage analysis and association analysis. Linkage studies involve genetic mapping based on the cotransmission of genetic markers and phenotypes from one generation to the next in one or more families. Association studies involve comparison of cases to controls to assess the relative contribution of genetic variants or environmental effects to the trait being studied. In addition, association studies may also be designed to study a quantitative trait, such as IOP, in a single large cohort.
Primary Glaucomas
Primary Congenital Glaucoma
Primary congenital glaucoma (PCG) is an uncommon disease with a frequency ranging from 1 in 1250 (among the Roma population of Slovakia) to 1 in 10,000 (2). The anterior segment often reveals an anteriorly inserted iris, with a maldeveloped angle and trabecular meshwork. Most cases of PCG are sporadic; in familial cases, autosomal-recessive inheritance is most common. Most of these patients require surgical management because current glaucoma medications and lasers are generally ineffective for this form of glaucoma. Two loci have been identified for the infantile form of congenital glaucoma: 2p211 and 1p36. The gene within the 2p21 locus, which accounts for the majority of familial cases, was identified in 1997 and encodes the protein cytochrome P4501B1 (CYP4501B1).
1Chromosomal location is numbered according to the following convention: The first number indicates the chromosome number, the letter “q” indicates long arm and “p” the short arm (petit in French), and the final number (with or without decimal point) indicates the band number on the chromosome; the banding is numbered according to distance from the centromere and morphologic features consistently found on the chromosome, such as Giemsa-staining band pattern.
Although the ocular substrate for cytochrome P450B1 remains unknown, this enzyme is likely to play an important role in ocular development (3). Libby and colleagues have shown that mutant Cyp1b1–/– mice deficient in cytochrome P450B1, where both copies of the Cyp1b1 gene are nonfunctional, develop focal defects in the anterior chamber angle, including an increase in basal lamina of the trabecular meshwork and a small or absent Schlemm canal. Other experiments testing for genes that enhance or suppress angle abnormalities in Cyp1b1 identified the tyrosinase gene (Tyr) as a modifier whose deficiency exacerbates defects in Cyp1b1 mutant mice (3). Eyes lacking cytochrome P450B1 and tyrosinase demonstrated severe dysgenesis that was alleviated by the administration of l-DOPA, a normal product of tyrosinase. Thus, a pathway involving tyrosinase appears to be important in anterior chamber angle development.
Juvenile-Onset Open-Angle Glaucoma
JOAG is an autosomal-dominant form of COAG with an early age of onset. It is characterized by extremely high IOP with subsequent damage to the optic nerve and visual field. Affected eyes are often myopic. This disease usually begins between the ages of 4 and 35 years, often in individuals with a strong family history. In patients with JOAG, response to drug or laser treatment is generally poor and surgical intervention is often required.
JOAG was first linked to chromosome 1q21–31 by Sheffield and colleagues in 1993. Four years later, mutations were found in the responsible gene, the trabecular meshwork glucocorticoid response gene (TIGR, later renamed myocilin (4)). At least five loci are now mapped for JOAG. Of all cases of JOAG, approximately 10% to 20% are caused by mutations in the myocilin gene (5).
Revisiting Case 1
The phenotype is classic for JOAG. A mutation in the myocilin gene was suspected, and hence the gene was sequenced. A single base change, C→T (Pro370Leu) in exon 3, was found (6). This missense mutation was found in the mother and the two affected daughters, but not in the father. Armed with this information, one can now respond to the patient’s queries:
- What do I have?
JOAG
- Will I go blind if I don’t receive treatment, and what is my best treatment option?
The Pro370Leu mutation is aggressive and leads to blindness if the pressure elevation is not treated. The best treatment option at present is aggressive IOP lowering with medication initially, and then surgery (e.g., trabeculectomy with an antimetabolite) if medical treatment does not lower the IOP to an appropriate target range.
- What are the chances that any future biologic children of mine would also get this disease?
JOAG is autosomal dominant with high penetrance, so the risk is approximately 50%.
- Can anything be done other than medications and surgery to treat my condition?
Not at present, but additional strategies may become possible in the future, including gene replacement and alteration of the trabecular meshwork cellular and extracellular milieu to enhance outflow facility.
Adult-Onset Chronic Open-Angle Glaucoma
The high prevalence of COAG, variability in age of onset, and nonpenetrance (lack of phenotypic expression of a disease despite carrying the genetic mutation) in some pedigrees indicate that most cases of COAG are not inherited as a single-gene defect but as a “complex” trait that does not demonstrate simple Mendelian inheritance. Interplay among various environmental and genetic factors, or among multiple genes, results in a high degree of variability in phenotypic expression and disease severity that makes linkage analysis extremely challenging. To date, linkage studies on families with COAG provide strong evidence for genetic heterogeneity. At least 11 loci have been identified, along with three genes (myocilin, optineurin, and WDR36) (Table 8.1).
Additional evidence for genetic susceptibility comes from polymorphisms of genes suspected of playing a role in glaucoma. Polymorphisms in the genes coding for the β-adrenergic receptors ADRB1 and ADRB2 expressed in the trabecular meshwork and ciliary body have been examined and may influence the pathophysiology of COAG in both COAG and normal-tension glaucoma in Japanese patients (7). However, the ADRB2 gene does not appear to be a “causative” COAG genetic risk, as shown in an appropriately powered study comparing controls and COAG cases among white individuals and persons of African ancestry (8). There may also be susceptibility genes that are essential to permit other genes or environmental factors to lead to glaucoma. For example, the OPA1 gene and apolipoprotein E gene have been associated with normal-tension glaucoma and COAG, respectively (9,10). It remains to be seen what role these disease-associated polymorphisms will play in patients with glaucoma.
Angle-Closure Glaucoma
There have been a growing number of investigators who have explored the familial basis of angle-closure glaucoma using both traditional Mendelian study design approaches and application of ocular biometry for quantitative trait design approach. In certain regions of the world, angle-closure glaucoma is the most common form of glaucoma, so it is important to understand the genetic mechanisms involved in this condition, which can be amenable to treatment with laser approaches.
Using a combination of a genetic approach applied to an epidemiology study, Hu found a sixfold-increased risk for angle-closure glaucoma among persons with any family history of angle-closure glaucoma in his population-based survey in Shunyi County, Beijing, which supports a genetic factor (11). Using a quantitative trait approach, a study of axial anterior chamber depth in twins (without angle-closure glaucoma) indicated that about 70% of the variance in dizygotic twins could be attributable to a genetic component (12). A biometric study showed a relatively shallow anterior chamber depth in siblings, children, nephews, nieces, and grandchildren of angle-closure glaucoma probands (13). A heritability of 70% was found in this study, indicating that about two thirds of the age- and sex-independent variation of anterior chamber depth is inherited. Furthermore, Lowe has suggested that inheritance of a shallow anterior chamber is polygenic with a threshold effect so that the action of a large number of grouped or independently inherited genes results in varying degrees of anterior chamber shallowing (14). A Chinese study of families with angle-closure glaucoma and shallow anterior chambers concluded that the inheritance of a shallow anterior chamber may be a genetically heterogeneous trait and influenced by sex with autosomal-dominant inheritance in subgroups (15).
In a rare phenotype on the spectrum of angle-closure glaucoma is nanophthalmos, which represents an ocular phenotype characterized by a biometrically small eye with relatively normal lens volume. Such individuals are at increased risk for angle-closure glaucoma due to a crowded anterior segment, uveal effusions due to thickened sclera, and aqueous misdirection (see Chapter 26). In a large family with 22 affected family members with highly penetrant nanophthalmos (16), a locus called NNO1 was mapped to chromosome 11. The gene has not yet been identified.
Using a molecular approach, a study quantifying SPARC protein (secreted protein, acidic, and rich in cysteine) in iridectomy specimens of eyes with chronic angle closure found that these irides had a significantly higher SPARC and collagen 1 protein content compared with nonglaucomatous eyes and eyes with COAG (17). The data suggest that SPARC could play a role in the development of angle-closure glaucoma by influencing the biomechanical properties of the iris through a change in extracellular matrix organization.
It has also been suggested that environmental triggers may alter anterior chamber depth or degree of pupillary block. These are associated with angle-closure glaucoma, including neural or humoral response to fatigue, mental stress, infection, and trauma (18).
Secondary Glaucomas
Developmental Glaucomas
Developmental glaucomas are secondary to morphologic malformations of the anterior segment and are relatively rare. Importantly, however, developmental abnormalities of the ocular drainage structures are not always clinically detectable, and abnormal development may affect the metabolism and function of the drainage structures without disturbing morphology. Glaucomas and known genes associated with developmental disorders are listed as part of Table 8.1. It is important to note that clinical findings overlap considerably, even within families, and mutations in the same gene can cause a range of phenotypes. The primary causative genes that have been identified are transcription factor–related genes: PITX2, PITX3, and FOXC1.
Pigmentary Glaucoma
Several investigators have demonstrated autosomal-dominant inheritance for the pigment dispersion syndrome (PDS) (19–21). In 1997, Andersen and colleagues described four autosomal-dominant PDS families and reported localization of a gene to chromosome 7q35–36 (22). The disorder is genetically heterogeneous, and further studies are under way to determine whether additional loci exist and to find the gene (or genes) involved. DBA/2J mice appear to develop a form of pigmentary glaucoma caused by mutations in the glycoprotein (transmembrane) nmb gene, Gpnmb, and the tyrosinase-related protein 1 gene, Tyrp1. As both genes encode melanosomal proteins, it has been hypothesized that these mutations permit toxic intermediates of pigment production to leak from melanosomes (23). A study examining glaucoma patients with PDS for DNA sequence variants in TYRP1 did not find an association (24).
Exfoliation Syndrome
Evidence supports the concept that exfoliation is an inherited microfibrillopathy involving transforming growth factor-1, oxidative stress, and impaired cellular protection mechanisms as key factors (Fig. 15.12). In a study in the Icelandic and Swedish populations, a common genetic variant was identified as a major risk factor for exfoliation syndrome and glaucoma (25). Polymorphisms in the coding region of the gene lysyl oxidase-like 1 (LOXL1), located on chromosome 15q24, are associated with exfoliation and exfoliative glaucoma in these and other populations. The disease-associated polymorphisms are found in virtually all individuals with exfoliation within populations studied to date.
LOXL1 is one of many enzymes essential for the formation of elastin fibers: It plays a role in modifying tropoelastin, the basic building block of elastin, and catalyzes the process for monomers to cross-link and form elastin. Although LOXL1 is a major risk factor for exfoliation syndrome and exfoliative glaucoma, evidence suggests that additional genetic or environmental factors will be identified that influence disease expression and severity. One example is a study of white persons in Australia with a ninefold-lower lifetime incidence of exfoliative glaucoma compared with Scandinavian populations that demonstrated a similar allelic architecture at the LOXL1 locus (26). This suggests that unidentified genetic or environmental factors independent of LOXL1 strongly influence the phenotypic expression of the syndrome.
The disease-associated LOXL1 variant is extremely common and is found in up to 90% of affected and unaffected individuals worldwide. For this reason, genetic testing is of limited clinical value at this time (27).
Revisiting Case 2
The discovery of the variants in the LOXL1 gene has the potential to lead to more exact diagnosis, better monitoring of glaucoma suspects, improved knowledge of pathogenesis, and eventually more effective treatment. Despite the importance of the identification of LOXL1 as a major contributor to exfoliation syndrome and exfoliative glaucoma, given the high frequency of disease-associated polymorphisms in the population, DNA testing is not clinically useful at this time.
Systemic Diseases Associated with Glaucoma
A number of ocular disorders that have been linked are associated with open-angle forms of glaucoma as part of their phenotype. These are listed in Table 8.1. In addition, a number of systemic disorders are associated with open-angle forms of glaucoma (e.g., nail–patella syndrome and Marfan syndrome), and those for which the gene has been localized or identified are listed in Table 8.1.
GENETICS AND INSIGHTS INTO DISEASE MECHANISMS
After identifying genes that are causative for glaucoma and genes that contribute to risk factors for glaucoma, we will elucidate disease mechanisms for glaucoma. This will also involve well-established mouse-model systems for glaucoma that will allow studies on specific biochemical pathways that ultimately cause glaucoma (28). To reach an in-depth understanding of role of these genes among these pathways, however, it will be essential to combine the tools of genomics, molecular biology, developmental biology, bioinformatics, and computational biology. This should ultimately lead to a better understanding of the normal physiology of the trabecular meshwork, optic nerve, ganglion cells, and other glaucoma-relevant tissues. Improved understanding of the state of the eye in disease and health will facilitate the rational development of drugs tailored to specific subtypes of glaucoma.
PHARMACOGENETICS, PHARMACOGENOMICS, AND THE PROMISE OF “PERSONALIZED MEDICINE”
Although all this information on genetics may appear daunting to the clinician, it is important to put this genomic technology in perspective. All of this genomic information, and the anticipated proteomic and metabolomic information, will not substitute for solid clinical history-taking skills, observation, assessment, and development of a treatment plan for the individual patient. However, at present, using our clinical acumen, our treatment approach is a trial-and-error approach by recommending a medication, laser, or surgery with an expected optimal treatment outcome. There is great optimism that genetic profiling will help target patients with glaucoma to individualized treatments on the basis of validated disease-risk alleles, validated pharmacogenetic markers, and specific behavioral modification. Thus, one may view these newer technology advances to take the guesswork out of the treatment plan, with the expectation of improved efficacy because the optimal treatment is specified for certain individual profiles and for decreased adverse events to treatment because it will not be recommended in a susceptible individual.
It is important to remember, however, that genes merely represent the blueprint to uncover genetic variants in common diseases, and they will not provide “the answer” to the question “What causes glaucoma?” Considerable strides are needed to fully understand factors that affect gene expression, such as DNA methylation, gene repair, copy-number variation, and telomerase action. In addition, proteomics is arguably just as crucial to genomics when looking at normal physiology and disease. For instance, posttranslational modifications, such as glycosylation, adenosine diphosphate–ribosylation, and phosphorylation, that affect cell function may also contribute to differences in an individual’s disease manifestation and response to treatment.
Pharmacogenomic studies could reveal genetic factors that predispose to poor IOP response (Fig. 8.6) as well as to higher-than-average risk for an adverse response—for example, the development of elevated IOP in response to corticosteroid therapy.
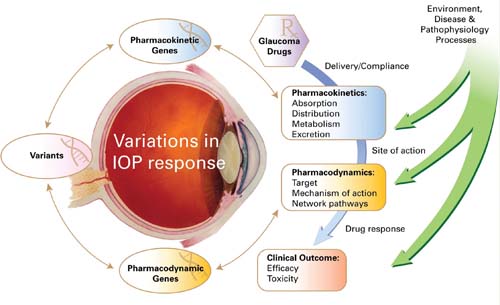
Figure 8.6 Variations in IOP response to glaucoma medical therapy are determined by pharmacokinetic and pharmacodynamic processes (blue arrow) and interaction with the environment, disease, and pathophysiologic processes. The sequence variants among pharmacokinetic and pharmacodynamic genes are predicted to have functional consequences that contribute to the genetic component of variance in IOP response. (Modified from pharmgkb.org, with permission of PharmGKB and Stanford University.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


