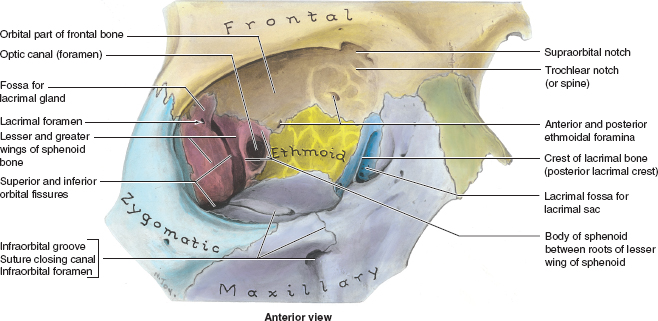
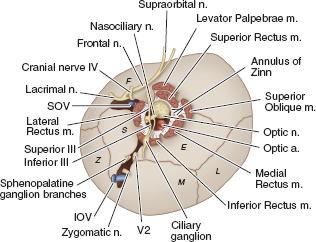
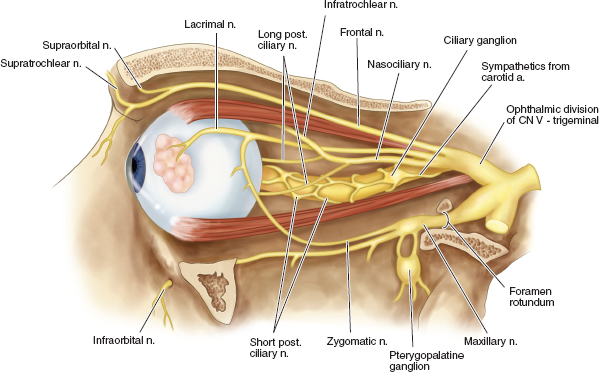
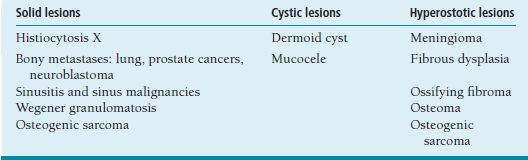
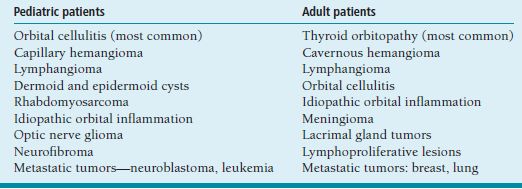
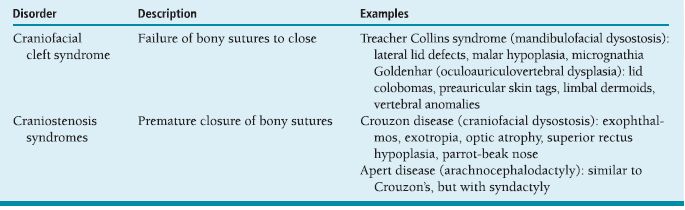
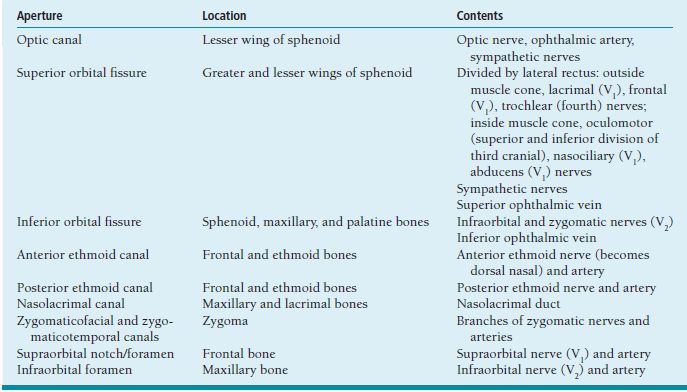
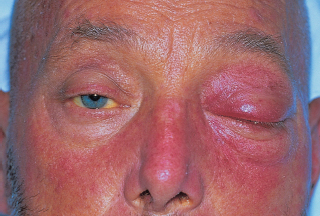
ORBIT Anatomy Orbital Dimensions Volume: 30 mL. Height: 35 mm. Width: 40 mm. Medial wall length: 45 mm. Globe to optic canal: 18 mm. Optic nerve: 5 cm. Intraocular: 1 mm. Intraorbital: 25 mm. Intracanalicular: 10 mm. Intracranial: 15 mm. Orbital Bones See Figure 7.1. Orbital Apertures See Figures 7.2 and 7.3, and Table 7.1. Evaluation of Orbital Disorders Axial displacement (Table 7.2). Nonaxial displacement (Table 7.3). Imaging (Table 7.4). Destructive lesions of bone (Table 7.5). Classification of orbital disorders (Tables 7.6 and 7.7). FIGURE 7.1. Seven orbital bones: sphenoid, ethmoid, zygomatic, maxillary, frontal, lacrimal, and palatine, with the ethmoid being the thinnest bone. Observe the optic canal situated within the lesser wing of the sphenoid bone, separated by optic strut from the superior orbital fissure. (From Moore KL, Dalley AF II. Clinical oriented anatomy, 4th ed. Baltimore, MD: Lippincott Williams & Wilkins, 1999, with permission.) FIGURE 7.2. The orbital apex. (E, ethmoid bone; F, frontal bone; IOV, inferior orbital vein; L, lacrimal bone; M, maxillary bone; S, sphenoid bone; SOV, superior orbital vein; Z, zygomatic bone.) Orbital Cellulitis Summary Bacterial and fungal infections of the orbit may occur in children (usually monomicrobial) and adults (polymicrobial) with vision-threatening and life-threatening sequelae. Preseptal cellulitis occurs only in the periorbital structures anterior to the orbital septum, unlike orbital cellulitis, which involves more posterior structures. Orbital cellulitis most commonly occurs in the setting of active paranasal sinus disease or trauma with associated infected facial wounds. Etiology Periorbital structures: paranasal sinus, face and eyelid, lacrimal sac (dacryocystitis), or dental infections. Intraorbital structures: endophthalmitis, dacryoadenitis. Exogenous: trauma, surgery. Endogenous: septic embolization. Most common organisms: Staphylococcus aureus, Streptococcus sp., Haemophilus influenzae (in children), Gram-negative rods, Mucor (in diabetics), Aspergillus. Signs and Symptoms Special Tests FIGURE 7.3. The nerves of the orbit. Observe the ciliary ganglion that receives sensory fibers from the nasociliary branches of CN V1, sympathetic fibers from the internal carotid plexus (traveling around the ophthalmic artery), and parasympathetic fibers (which synapse in the ganglion) from the inferior branch of the CN III. Table 7.2 Axial displacement aAll causes listed below may cause unilateral proptosis as well. Table 7.3 Nonaxial displacement Table 7.5 Destructive lesions of bone Table 7.6 Common orbital disorders Table 7.7 Craniofacial deformities Mucormycosis Disease Course Table 7.8 Signs and symptoms of orbital cellulitis Treatment and Management Medications Antibiotic choice depends on origin or cause of cellulitis. Follow-up Differential Diagnosis References Bilyk J. Periocular infection. Curr Opin Ophthalmol 2007;18: 414–423. Harris GJ. Subperiosteal abscess of the orbit. Age as a factor in the bacteriology and response to treatment. Ophthalmology 1994;101(3):585–595. Necrotizing Fasciitis Summary Etiology Signs and Symptoms Treatment and Management Reference Elner VM, Demirci H, Nerad JA, et al. Periocular necrotizing fasciitis with visual loss. Ophthalmology 2006;113:2338–2345. Thyroid Orbitopathy Summary Thyroid orbitopathy, the most common cause of unilateral or bilateral proptosis in adults, occurs in 50% of patients with Graves disease. Patients with thyroid orbitopathy may be hyperthyroid, hypothyroid, or even euthyroid. Ocular complications attributable to thyroid orbitopathy include ocular exposure, strabismus, and optic nerve compression. Etiology Signs and Symptoms See Figure 7.5. Demographics Ophthalmic Findings Werner’s “NOSPECS” (mnemonic) classification: Class 0: No signs or symptoms. Class 1: Only signs (eyelid retraction). Class 2: Soft tissue periorbital swelling. Class 3: Proptosis of eyes. Class 4: Extraocular muscle involvement, diplopia. Class 5: Corneal involvement, exposure. Class 6: Sight loss by optic nerve compression. Systemic Findings Special Tests Extraocular muscle enlargement sparing tendons: Most commonly involved (in order): inferior rectus, medial rectus, and superior rectus muscles. Least commonly involved: lateral rectus. Increased fat volume. Optic nerve compression by muscles in orbital apex. Rule out other causes of proptosis. Define anatomy of paranasal sinuses before orbital decompression with CT scan. Pathology Inflammation and enlargement of orbital tissues, specifically the nontendinous portions of the extraocular muscles: Early: lymphocyte and plasma cell infiltration of muscles, deposition of mucopolysaccharide by activated fibroblasts, and formation of collagen. Late: fibrosis and fatty infiltration of muscles. Disease Course Treatment and Management For acute inflammatory phase. For compressive optic neuropathy. Prednisone 60 to 100 mg for up to 1 week to assess steroid responsiveness. Pulse IV methylprednisolone (10 mg/kg, max 1 g) repeat at 48-hour intervals × 3; may be repeated after 3 months if no response or recurs. Side effects of cardiac arrhythmia, liver toxicity, relapse. Rebound effect may occur after cessation of steroids. Cyclosporin A: inhibits T-cell and IL-2 activity; start 2 mg/kg BID. Tacrolimus: inhibits T-cell and IL-2 activity. Traditionally recommended for acute inflammatory phase or compressive optic neuropathy; however, recent studies have failed to demonstrate significant benefits 12 months after radiotherapy, whether administered early or late in the course of Graves ophthalmopathy. Risks include cataract formation and radiation retinopathy, especially in diabetics and patients receiving systemic chemotherapy. For compressive optic neuropathy: dosage: 2,000 cGy in ten fractions over 2 weeks to posterior orbit. Orbital decompression. Extraocular muscle surgery. Adjustment of eyelid margin position. Blepharoplasty. For optic neuropathy, corneal exposure, proptosis. Remove bony walls of orbit to allow prolapse of orbital soft tissues and enlargement of orbital volume. Medial wall, ethmoid sinus, orbital floor, maxillary sinus, lateral wall, temporalis fossa. Good success rates in improving decreased vision attributable to optic neuropathy. Complications: diplopia, hemorrhage, infection, cerebrospinal fluid leak, optic nerve injury. Perform only after motility disturbance is stable for 6 months. Steroids and radiation therapy have minimal effect. Inferior and medial rectus muscles most often affected: recess with adjustable sutures. Lid retraction due in part to fibrotic levator or Müller muscle, increase in sympathetic activity. Recess either muscle to lower the lid, especially laterally, with or without spacer grafts such as sclera. Raise the lower lid to help cover the cornea, with spacer grafts. Differential Diagnosis Idiopathic orbital inflammation (pseudotumor). Orbital amyloidosis. Carotid cavernous fistula. Orbital tumor. Eyelid retraction: see p. 29 for differential. References Gorman CA, Garrity JA, Fatourechi V, et al. A prospective, randomized, double-blind, placebo-controlled study of orbital radiotherapy for Grave’s ophthalmopathy. Ophthalmology 2001;108:1523–1534. Krassas GE, Boboridis K. Recent developments in the medical treatment of thyroid eye disease. Orbit 2006;25:117–122. Meyer PAR. Avoiding surgery for thyroid eye disease. Eye 2006;20:1171–1177. Sergott RC, Glaser JS. Graves’ ophthalmopathy: a clinical and immunologic review. Surv Ophthalmol 1981;26:1–21. Shorr N, Seiff SR. The four stages of surgical rehabilitation of the patient with dysthyroid ophthalmopathy. Ophthalmology 1986;93:476–483. Idiopathic Orbital Inflammation (Orbital Pseudotumor) Summary Acute idiopathic orbital inflammation typically presents with pain and may be diffuse or localized to a specific area of the orbit. Treatment is typically with oral steroids. Many systemic processes such as sarcoid, Wegener granulomatosis, or metastatic carcinomas may present in a similar fashion. These must be considered before the diagnosis of idiopathic orbital inflammation is made. Etiology Idiopathic. Signs and Symptoms Pain. Erythema and edema of lids. Chemosis. Decreased vision. Decreased extraocular motility. Uveitis. Pain. Decreased vision. Decreased extraocular motility. Pain with eye movements. Decreased extraocular motility. Localized chemosis. Pain. S-shaped eyelid deformity. Localized chemosis. Demographics All ages, men and women. Ophthalmic Findings Children may develop papillitis. Special Tests CT scan: Anterior, diffuse, apical: enhancing infiltrative mass with irregular margins. Myositic: fusiform enlargement of extraocular muscle involving the tendon. Lacrimal: irregular swelling of lacrimal gland. Pathology Polymorphous infiltrate of inflammatory cells including lymphocytes, neutrophils, plasma cells, and macrophages. Disease Course Generally resolves with treatment but may recur. Treatment and Management Start with 1.0 to 1.5 mg/kg/day × 1 to 2 weeks, with slow taper over 6 to 12 weeks. Methylprednisolone 1 g/day × 1 to 3 days for vision loss Rapid resolution usually occurs with steroids. Differential Diagnosis Orbital cellulitis. Orbital inflammation secondary to the following: Sarcoidosis: Uveitis, dacryoadenitis, rare orbital lesions. Tests: angiotensin-converting enzyme, lysozyme, chest x-ray (CXR): hilar adenopathy, positive gallium scan. Pathology: noncaseating granulomas. Wegener granulomatosis: Upper and lower respiratory tract lesions, vasculitis, nephritis, orbital inflammation, uveitis. Tests: antineutrophil cytoplasmic antibodies (ANCA), abnormal urinary sediment, abnormal CXR. Pathology: necrotizing granulomatous inflammation and vasculitis Sjögren syndrome: Dry eyes, dry mouth, other associated autoimmune diseases, enlarged lacrimal glands. Tests: Antinuclear antibodies (ANA, rheumatoid factor [RF], Sjögren antibodies). Pathology: polymorphous nongranulomatous inflammatory infiltrate Ruptured dermoid cyst. Arteriovenous fistulas. Orbital metastasis. Orbital cellulitis. Ruptured dermoid cyst. Rhabdomyosarcoma. Metastatic neuroblastoma. Leukemic orbital infiltrate. References Harris GJ. Idiopathic orbital inflammation: a pathogenetic construct and treatment strategy. Ophthal Plast Reconstr Surg 2006;22:79–86. Mombaerts I, Glodschmeding R, Schlingemann RO, et al. What is orbital pseudotumor? Surv Ophthalmol 1996;41:66–78. Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982;89:1040–1048. Congenital Orbital Tumors Dermoid and Epidermoid Cysts Clinical Features Pathology Treatment Lipodermoid Clinical Features Pathology Solid tumor with adipose tissue, hair follicles. Treatment Reference Fry CL, Leone CR Jr. Safe management of dermolipomas. Arch Ophthalmol 1994;112:1114–1116. Vascular Tumors See Table 7.10. References Harris GJ. Orbital vascular malformations: a consensus statement on terminology and its clinical implications. Am J Ophthalmol 1999;127:453–455. Rootman J. Vascular malformations of the orbit: hemodynamic concepts. Orbit 2003;22:103–120. Capillary Hemangioma Clinical Features Pathology Fine network of endothelially lined vascular channels. Treatment CT, computed tomography Intralesional (betamethasone and triamcinolone): risk of emboli of steroid particles through vessels that anastomose with the orbital circulation and cause central retinal artery occlusion. Topical (clobetasol [Temovate] steroid cream): risk of atrophy and hypopigmentation of skin and subcutaneous tissues. Systemic oral steroids. Watch for adrenal suppression and growth retardation with steroids. Effective treatment for disfiguring infantile capillary hemangiomas. Potential mechanisms of action: vasoconstriction, down-regulation of the RAF/mitogen-activated protein kinase pathway resulting in decreased expression of proangiogenic factors, and triggering apoptosis of capillary endothelial cells. Risk of bradycardia, hypotension, hypoglycemia, bronchospasm. References Shorr N, Seiff SR. Central retinal artery occlusion associated with periocular corticosteroid injection for juvenile hemangioma. Ophthalmic Surg 1986;17:229–231. O’Keefe M, Lanigan B, Byrne SA. Capillary haemangioma of the eyelids and orbit: a clinical review of the safety and efficacy of intralesional steroid. Acta Ophthalmol Scand 2003;81: 294–298. Leaute-Labreze C, Dumas de la Roque E, Hubiche T, et al. Propranolol for severe hemangiomas of infancy. N Engl J Med 2008;358:2649–2651. Lymphangioma Clinical Features Pathology Treatment Differential Diagnosis Reference Harris GJ, Sakol PJ, Bonavolonta G, et al. An analysis of thirty cases of orbital lymphangioma: pathophysiologic considerations and management recommendations. Ophthalmology 1990;97:1583–1592. Cavernous Hemangioma Clinical Features Pathology Encapsulated lesion consisting of large, dilated vascular spaces filled with red blood cells. Treatment Surgical excision for lesions compromising ocular function. Hemangiopericytoma Clinical Features Pathology Large pericytes surround a capillary network. Treatment Neural Tumors Optic Nerve Glioma See Figure 7.8. Clinical Features Plain film: enlargement of optic canal greater than 6.5 mm in diameter. CT: S-shaped fusiform enlargement of optic nerve. Magnetic resonance imaging (MRI): better than CT for evaluation of posterior extension of tumor (more common in NF). Isointense or hypointense on T1 and hyperintense on T2. Pathology Pilocytic astrocytoma (hairlike elongated neoplastic glial cells) with Rosenthal fibers (eosinophilic cell processes). Treatment References Lee AG. Neuroophthalmological management of optic pathway gliomas. Neurosurg Focus 2007;23(5):E1. Liu GT. Optic gliomas of the anterior visual pathway. Curr Opin Ophthalmol 2006;17:427–431. Meningioma Clinical Features More common than primary optic nerve meningioma and can invade the orbit resulting in secondary optic nerve sheath meningioma. Middle-aged adult women. Sphenoid bone most commonly involved. Proptosis, chemosis, lid edema, temporal fullness, late visual loss. CT: bony change, hyperostosis. Middle-aged adult women; less commonly, children with neurofibromatosis. Proptosis, decreased vision, papilledema or optic atrophy, optociliary shunt vessels (collaterals from retinal to choroidal venous circulation; seen in 25% to 30%), choroidal folds. CT/MRI: diffuse thickening or fusiform enlargement of optic nerve, nodular surface, “train track” sign (central lucent zone), intralesional calcification. Pathology Treatment Excise for vision loss, motility disturbance, or ocular exposure. Combined approach with neurosurgery for surgical resection or debulking. More conservative treatment of optic nerve than intracranial meningioma, because of tumor location. Tumor is resected with risk of intracranial tumor spread, progressive growth, or poor vision. Complete surgical excision of optic nerve is performed with a blind eye and progressive tumor. Stereotactic radiotherapy is considered an early treatment option to stabilize and even improve vision. References Dutton JJ. Optic nerve sheath meningioma. Surv Ophthalmol 1992;37:167–183. Jeremic B, Pitz S. Primary optic nerve sheath meningioma. Cancer 2007;110:714–722. Plexiform Neurofibromas Clinical Features Pathology Intertwining bundles of Schwann cells, axons, and endoneural fibroblasts Treatment Debulk lesions with caution. Schwannoma Clinical Features Treatment Excision. Neuroblastoma Summary Mesenchymal Tumors Rhabdomyosarcoma Summary Rhabdomyosarcoma, the most common primary orbital malignancy of childhood, should be suspected in any child with the rapid onset of an orbital process. Biopsy of a suspected lesion should be performed immediately. The survival rate of patients with rhabdomyosarcoma has improved significantly with the use of chemotherapy and radiation therapy. Signs and Symptoms Demographics Onset: age 7 to 8 years. Ophthalmic Findings Systemic Findings Regional lymphadenopathy with metastatic spread to lung and bone. Special Tests Pathology Most common type. Superonasal quadrant of the orbit. Loose fascicles of undifferentiated spindle cells with rare cross striations. Most malignant form. Inferior orbit. Fibrovascular strands form alveolar-like compartments, which are lined with rhabdomyoblasts. Rarest form. Occurs in adults. Well differentiated with easily located cross striations. Variant of embryonal that occurs in grapelike clusters with invasion from the paranasal sinuses or conjunctiva. Disease Course Treatment and Management Differential Diagnosis Orbital cellulitis. Idiopathic orbital inflammation. Neuroblastoma. Lymphangioma with hemorrhage. Capillary hemangioma. Ruptured dermoid cyst. References Shields JA, Shields CL. Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol 2003;48:39–57. Wharam M, Beltangady M, Hays D, et al. Localized orbital rhabdomyosarcoma: an interim report of the intergroup rhabdomyosarcoma study committee. Ophthalmology 1987;94:251–254. Clinical Features Most common adult mesenchymal tumor of the orbit. Middle-aged adults. Proptosis, decreased vision. Range of benign to malignant based on clinical behavior. Local recurrence, rare metastasis. CT: well-circumscribed homogeneous mass (similar to cavernous hemangioma or schwannoma). Pathology Fibroblasts and histiocytes in cartwheel or storiform pattern; diagnosis of exclusion after immunohistochemistry rules out other spindle cell tumors such as solitary fibrous tumor (CD34), melanoma (S100, HMB-45), and leiomyoma (smooth muscle antigen, desmin). Treatment Complete surgical resection or irradiation (malignant fibrous histiocytoma). Reference Shields JA, Shields CL. Orbital fibrous histiocytoma. In: Shields JA, ed. Eyelid, conjunctival, and orbital tumors, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2008:626–631. Solitary Fibrous Tumor Clinical Features Pathology Treatment Reference Bernardini FP, Conciliis C, Schneider S, et al. Solitary fibrous tumor of the orbit. Ophthalmology 2003;110:1442–1448. Fibro-Osseous Tumors of the Orbit See Table 7.11. Reference Selva D, White VA, O’Connell JX, et al. Primary bone tumors of the orbit. Surv Ophthalmol 2004;49:328–342. Histiocytic Tumors Langerhans Cell Histiocytosis (Histiocytosis X) Clinical Features Pathology Treatment Surgical curettage, systemic or local corticosteroids, local irradiation, cytotoxic agents. Reference Margo CE, Goldman DR. Langerhans Cell Histiocytosis. Surv Ophthalmol 2008;53:332–358. Ocular Adnexal Lymphoid Tumors Summary Lymphoproliferative lesions occur as a continuum from benign lymphoid hyperplasia (polyclonal) to malignant lymphoma (monoclonal) and should be regarded as a potentially systemic process. Orbital lymphoma is the most common malignant orbital tumor affecting older adults. Signs and Symptoms Demographics Sixth and seventh decades of life. Rare in children. Systemic Findings Risk of developing systemic non-Hodgkin lymphoma based on anatomic site of origin (lowest to highest): conjunctival (10%), orbital (50%), and eyelid (66%).
 Cultures and/or Gram stain: blood, conjunctiva, nasopharynx, external wounds, cerebrospinal fluid, as necessary.
Cultures and/or Gram stain: blood, conjunctiva, nasopharynx, external wounds, cerebrospinal fluid, as necessary.
 Complete blood cell count (CBC) with differential.
Complete blood cell count (CBC) with differential.


 Computed tomography (CT) scan: Order with contrast, evaluate preseptal versus postseptal location, look for paranasal sinus disease, subperiosteal abscess.
Computed tomography (CT) scan: Order with contrast, evaluate preseptal versus postseptal location, look for paranasal sinus disease, subperiosteal abscess.
 Biopsy for microscopy.
Biopsy for microscopy.
 Fungus: Mucor—nonseptate hyphae, Aspergillus—septate branching hyphae.
Fungus: Mucor—nonseptate hyphae, Aspergillus—septate branching hyphae.
 Severe infection with genera Rhizopus, Mucor, Absidia belonging to the family Mucoraceae, a subset of the class Phycomycetes.
Severe infection with genera Rhizopus, Mucor, Absidia belonging to the family Mucoraceae, a subset of the class Phycomycetes.
 Blood vessel invasion, vascular occlusion, necrosis, black eschar (late finding) on roof of mouth or nose.
Blood vessel invasion, vascular occlusion, necrosis, black eschar (late finding) on roof of mouth or nose.
 Begins in sinus—invades orbit.
Begins in sinus—invades orbit.
 Risk factors: diabetics in ketoacidosis or with antecedent bacterial infection, alcoholics, and immunocompromised patients.
Risk factors: diabetics in ketoacidosis or with antecedent bacterial infection, alcoholics, and immunocompromised patients.
 Histopathology: nonseptate, large branching filamentous hyphae seen on periodic acid-Schiff and Gomori methenamine-silver stains.
Histopathology: nonseptate, large branching filamentous hyphae seen on periodic acid-Schiff and Gomori methenamine-silver stains.
 Treatment: extensive debridement of necrotic tissues with irrigation with amphotericin B.
Treatment: extensive debridement of necrotic tissues with irrigation with amphotericin B.
 Intravenous amphotericin B (nephrotoxic, aplastic anemia).
Intravenous amphotericin B (nephrotoxic, aplastic anemia).
 May progress from preseptal to orbital cellulitis, orbital cellulitis to cavernous sinus thrombosis or intracranial abscess, without appropriate treatment. Must follow patients closely for development of deeper infection, decreased vision or ocular motility, or afferent pupillary defect.
May progress from preseptal to orbital cellulitis, orbital cellulitis to cavernous sinus thrombosis or intracranial abscess, without appropriate treatment. Must follow patients closely for development of deeper infection, decreased vision or ocular motility, or afferent pupillary defect.
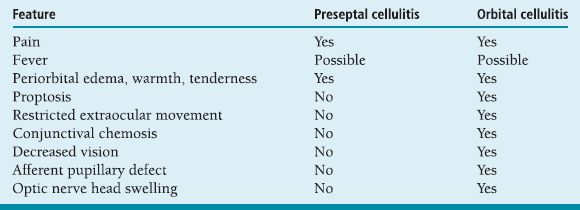
 Cavernous sinus thrombosis: bilateral orbital involvement, decreased skin sensation affecting ophthalmic (V1) or maxillary (V2) divisions of the trigeminal nerve, complete ophthalmoplegia, headache, nausea, vomiting, and fever.
Cavernous sinus thrombosis: bilateral orbital involvement, decreased skin sensation affecting ophthalmic (V1) or maxillary (V2) divisions of the trigeminal nerve, complete ophthalmoplegia, headache, nausea, vomiting, and fever.
 Subperiosteal abscess formation (commonly superomedial or inferomedial orbit) can result in sudden loss of vision with posterior extension of the abscess toward the orbital apex.
Subperiosteal abscess formation (commonly superomedial or inferomedial orbit) can result in sudden loss of vision with posterior extension of the abscess toward the orbital apex.
 After cultures and blood work are obtained, the patient should be immediately placed on a regimen of antibiotics. Infectious disease consultation may be obtained.
After cultures and blood work are obtained, the patient should be immediately placed on a regimen of antibiotics. Infectious disease consultation may be obtained.
 Patients with preseptal cellulitis are occasionally treated as outpatients with oral antibiotics; otherwise, intravenous antibiotics are given.
Patients with preseptal cellulitis are occasionally treated as outpatients with oral antibiotics; otherwise, intravenous antibiotics are given.
 Patients with sinus disease should be evaluated by an otolaryngologist.
Patients with sinus disease should be evaluated by an otolaryngologist.
 Mucormycosis, a rare, rapidly progressive fungal orbital infection, should be suspected in all diabetics and immunosuppressed patients; definitive diagnosis and treatment should be performed immediately; treatment consists of orbital debridement, possible exenteration and local irrigation, and systemic treatment with amphotericin B.
Mucormycosis, a rare, rapidly progressive fungal orbital infection, should be suspected in all diabetics and immunosuppressed patients; definitive diagnosis and treatment should be performed immediately; treatment consists of orbital debridement, possible exenteration and local irrigation, and systemic treatment with amphotericin B.
 A lag time of 24 to 48 hours commonly occurs between initiation of antibiotics and clinical response.
A lag time of 24 to 48 hours commonly occurs between initiation of antibiotics and clinical response.
 If there is no improvement or if new signs such as decreased vision or development of an afferent pupillary defect develop, repeat CT scan and suspect abscess formation or resistant organisms.
If there is no improvement or if new signs such as decreased vision or development of an afferent pupillary defect develop, repeat CT scan and suspect abscess formation or resistant organisms.
 Abscesses in adults should be surgically drained, particularly in the setting of visual compromise; abscesses in children may be followed closely if under the age of 9, with surgical drainage for any deterioration.
Abscesses in adults should be surgically drained, particularly in the setting of visual compromise; abscesses in children may be followed closely if under the age of 9, with surgical drainage for any deterioration.
 Aspergillosis, a fungal infection that may be acute or chronic and local or disseminated, is treated with orbital debridement, possible exenteration and local irrigation, and systemic treatment with amphotericin.
Aspergillosis, a fungal infection that may be acute or chronic and local or disseminated, is treated with orbital debridement, possible exenteration and local irrigation, and systemic treatment with amphotericin.
 Trauma: third-generation cephalosporin and clindamycin for Gram negative organisms Gram-positive skin flora, and anaerobes, respectively.
Trauma: third-generation cephalosporin and clindamycin for Gram negative organisms Gram-positive skin flora, and anaerobes, respectively.
 Sinus: second-generation cephalosporin for sinus bacteria.
Sinus: second-generation cephalosporin for sinus bacteria.
 Children: single aerobe, second-generation cephalosporin for H. influenzae.
Children: single aerobe, second-generation cephalosporin for H. influenzae.
 Adults (>14 y/o): polymicrobial (average five species). Third-generation cephalosporin, nafcillin, or vancomycin (if methicillin-resistant S. aureus suspected), and metronidazole (crosses blood–brain barrier) or clindamycin for anaerobes.
Adults (>14 y/o): polymicrobial (average five species). Third-generation cephalosporin, nafcillin, or vancomycin (if methicillin-resistant S. aureus suspected), and metronidazole (crosses blood–brain barrier) or clindamycin for anaerobes.
 Fungus: amphotericin B (liposomal amphotericin B reserved for immunocompromised host with renal failure), voriconazole (more effective for invasive Aspergillus).
Fungus: amphotericin B (liposomal amphotericin B reserved for immunocompromised host with renal failure), voriconazole (more effective for invasive Aspergillus).
 Patients should be followed extremely closely until signs of improvement are evident, such as decreased lid edema and erythema, decreased chemosis, improved ocular motility, improved visual acuity, or decreased proptosis.
Patients should be followed extremely closely until signs of improvement are evident, such as decreased lid edema and erythema, decreased chemosis, improved ocular motility, improved visual acuity, or decreased proptosis.
 Intravenous antibiotics should be continued for approximately 1 week before switching to oral antibiotics.
Intravenous antibiotics should be continued for approximately 1 week before switching to oral antibiotics.
 Idiopathic orbital inflammation: no signs of infection.
Idiopathic orbital inflammation: no signs of infection.
 Carotid cavernous fistula: no signs of infection.
Carotid cavernous fistula: no signs of infection.
 Orbital vasculitis.
Orbital vasculitis.
 Periocular necrotizing fasciitis is a rapidly progressive infection that spreads along the subcutaneous soft tissue planes and that can eventually lead to shock and organ failure.
Periocular necrotizing fasciitis is a rapidly progressive infection that spreads along the subcutaneous soft tissue planes and that can eventually lead to shock and organ failure.
 Idiopathic but has been associated with blunt or surgical trauma (blepharoplasty) in healthy patients.
Idiopathic but has been associated with blunt or surgical trauma (blepharoplasty) in healthy patients.
 Group β-hemolytic Streptococcus.
Group β-hemolytic Streptococcus.
 Mimics preseptal cellulitis initially but results in necrosis of adjacent tissue and cyanosis (subcutaneous hemorrhage and thrombosis).
Mimics preseptal cellulitis initially but results in necrosis of adjacent tissue and cyanosis (subcutaneous hemorrhage and thrombosis).
 Painful, erythematous, and/or blue eyelids.
Painful, erythematous, and/or blue eyelids.
 Possible retinal artery occlusion due to thrombosis or ischemic necrosis of the deep orbit.
Possible retinal artery occlusion due to thrombosis or ischemic necrosis of the deep orbit.
 Subcutaneous debridement, resection of necrotic skin, and β-lactam antibiotics (penicillin, ceftriaxone, or clindamycin).
Subcutaneous debridement, resection of necrotic skin, and β-lactam antibiotics (penicillin, ceftriaxone, or clindamycin).
 Autoimmune disorder with antibodies to orbital fibroblasts demonstrated in some patients.
Autoimmune disorder with antibodies to orbital fibroblasts demonstrated in some patients.
 Fibroblasts can transform into adipocytes.
Fibroblasts can transform into adipocytes.
 Stimulation of glycosaminoglycans or collagen production.
Stimulation of glycosaminoglycans or collagen production.
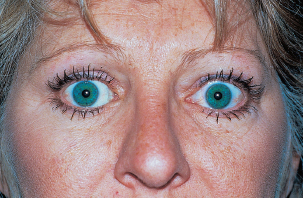
 Lid retraction (early sign).
Lid retraction (early sign).
 Lid lag on downgaze (early sign).
Lid lag on downgaze (early sign).
 Periorbital edema.
Periorbital edema.
 Proptosis.
Proptosis.
 Conjunctival chemosis.
Conjunctival chemosis.
 Enlarged vessels over rectus muscle insertions.
Enlarged vessels over rectus muscle insertions.
 Corneal exposure, ulceration.
Corneal exposure, ulceration.
 Extraocular motility restriction, with preferential extraocular muscle involvement (inferior > medial > superior > lateral rectus) sparing the tendons.
Extraocular motility restriction, with preferential extraocular muscle involvement (inferior > medial > superior > lateral rectus) sparing the tendons.
 Elevated intraocular pressure with upgaze.
Elevated intraocular pressure with upgaze.
 Compressive optic neuropathy with decreased visual acuity, decreased color vision, afferent pupillary defect, visual field defects.
Compressive optic neuropathy with decreased visual acuity, decreased color vision, afferent pupillary defect, visual field defects.
 Age: 25 to 50 years.
Age: 25 to 50 years.
 Ratio of women to men: 6 to 1.
Ratio of women to men: 6 to 1.
 Pretibial dermopathy.
Pretibial dermopathy.
 Other features associated with a dysthyroid state: restlessness, tremor, weight loss with hyperthyroidism or lethargy, constipation, weight gain with hypothyroidism.
Other features associated with a dysthyroid state: restlessness, tremor, weight loss with hyperthyroidism or lethargy, constipation, weight gain with hypothyroidism.
 Associated with myasthenia gravis, smoking, family history.
Associated with myasthenia gravis, smoking, family history.
 Evaluate thyroid function: sensitive thyroid-stimulating hormone (TSH); T3 and T4 (thyrotropin receptor) antibodies; antithyroid antibodies; thyrotropin-releasing hormone (TRH) stimulation test; Werner or T3 suppression test.
Evaluate thyroid function: sensitive thyroid-stimulating hormone (TSH); T3 and T4 (thyrotropin receptor) antibodies; antithyroid antibodies; thyrotropin-releasing hormone (TRH) stimulation test; Werner or T3 suppression test.
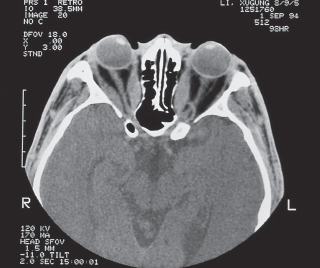
 Orbital imaging (Figure 7.6).
Orbital imaging (Figure 7.6).
 Acute inflammatory phase (6 to 18 months) evolves into chronic stable fibrotic phase.
Acute inflammatory phase (6 to 18 months) evolves into chronic stable fibrotic phase.
 Spontaneous remissions and exacerbations can occur.
Spontaneous remissions and exacerbations can occur.
 Thyroid disease and orbital disease run independent courses; however, orbital disease may worsen after radioactive iodide thyroid gland treatment.
Thyroid disease and orbital disease run independent courses; however, orbital disease may worsen after radioactive iodide thyroid gland treatment.
 Optic neuropathy may occur in patients without significant proptosis, because of posterior compression of the nerve.
Optic neuropathy may occur in patients without significant proptosis, because of posterior compression of the nerve.
 Fifty percent of patients with upper lid retraction may improve without intervention.
Fifty percent of patients with upper lid retraction may improve without intervention.
 Motility fluctuates during the acute phase.
Motility fluctuates during the acute phase.
 Local supportive measures: artificial teardrops, lubricating ointment, elevating head of bed.
Local supportive measures: artificial teardrops, lubricating ointment, elevating head of bed.
 Corticosteroids: given with H2 receptor antagonist, bisphosphonate, and calcium.
Corticosteroids: given with H2 receptor antagonist, bisphosphonate, and calcium.
 Other immunomodulators are being studied in refractory cases with active inflammation.
Other immunomodulators are being studied in refractory cases with active inflammation.
 Orbital radiation:
Orbital radiation:
 Surgical management: a staged approach must be taken to the surgical rehabilitation of the patient with thyroid orbitopathy so as to avoid unnecessary procedures.
Surgical management: a staged approach must be taken to the surgical rehabilitation of the patient with thyroid orbitopathy so as to avoid unnecessary procedures.
 Orbital decompression:
Orbital decompression:
 Extraocular muscle surgery:
Extraocular muscle surgery:
 Lid margin position surgery.
Lid margin position surgery.
 Blepharoplasty: Remove excess skin and fat to improve cosmesis.
Blepharoplasty: Remove excess skin and fat to improve cosmesis.
 Anterior orbit inflammation/diffuse inflammation:
Anterior orbit inflammation/diffuse inflammation:
 Apical inflammation:
Apical inflammation:
 Myositic inflammation:
Myositic inflammation:
 Lacrimal inflammation:
Lacrimal inflammation:
 Obtain orbital scan.
Obtain orbital scan.
 NSAIDS for myositis.
NSAIDS for myositis.
 Corticosteroids: first line in a typical case.
Corticosteroids: first line in a typical case.
 Biopsy if no improvement with steroids or if recurrence occurs, or atypical presentation.
Biopsy if no improvement with steroids or if recurrence occurs, or atypical presentation.
 Add immunosuppressants as steroid-sparing drugs.
Add immunosuppressants as steroid-sparing drugs.
 Orbital irradiation or immunobiologic agents (infliximab) can be used in refractory cases.
Orbital irradiation or immunobiologic agents (infliximab) can be used in refractory cases.
 Adults:
Adults:
 Children:
Children:
 Onset: childhood.
Onset: childhood.
 Smooth cystic mass in lateral or superolateral orbit.
Smooth cystic mass in lateral or superolateral orbit.
 Differentiate superomedial lesion from encephalocele by CT scan.
Differentiate superomedial lesion from encephalocele by CT scan.
 Dermoid: cyst lined by keratinized epidermis with dermal appendages such as hair follicles and sebaceous glands.
Dermoid: cyst lined by keratinized epidermis with dermal appendages such as hair follicles and sebaceous glands.
 Epidermoid: cyst lined by keratinized epidermis without dermal structures.
Epidermoid: cyst lined by keratinized epidermis without dermal structures.
 Complete excision.
Complete excision.
 Ruptured cyst results in severe inflammation.
Ruptured cyst results in severe inflammation.
 Solid choristoma located lateral to lateral limbus with orbital extension.
Solid choristoma located lateral to lateral limbus with orbital extension.
 Associated with Goldenhar syndrome.
Associated with Goldenhar syndrome.
 Excise anterior component only if hairs cause irritation.
Excise anterior component only if hairs cause irritation.
 Extensive resection may result in motility problems and damage to lacrimal ductules.
Extensive resection may result in motility problems and damage to lacrimal ductules.
 Onset at few weeks of life, grows over first year, involutes over several years.
Onset at few weeks of life, grows over first year, involutes over several years.
 Superficial strawberry surface lesion or deep bluish orbital lesion.
Superficial strawberry surface lesion or deep bluish orbital lesion.
 Kasabach-Merritt syndrome: capillary hemangiomas associated with thrombocytopenia.
Kasabach-Merritt syndrome: capillary hemangiomas associated with thrombocytopenia.
 Amblyopia, strabismus, anisometropia: treat; otherwise, may observe, since involution will occur.
Amblyopia, strabismus, anisometropia: treat; otherwise, may observe, since involution will occur.
 Steroids:
Steroids:
 Propranolol, systemic
Propranolol, systemic
 Surgery: useful only in well-circumscribed deep lesions.
Surgery: useful only in well-circumscribed deep lesions.
 Infiltrative nonencapsulated lesion of eyelid, orbit with onset in first decade
Infiltrative nonencapsulated lesion of eyelid, orbit with onset in first decade
 Abrupt enlargement with hemorrhage into lymphangioma (chocolate cyst)
Abrupt enlargement with hemorrhage into lymphangioma (chocolate cyst)
 May enlarge with upper respiratory tract infection.
May enlarge with upper respiratory tract infection.
 Fluid levels on MRI, but no flow on MRA or MRV.
Fluid levels on MRI, but no flow on MRA or MRV.
 Large serum-filled spaces lined by endothelial cells.
Large serum-filled spaces lined by endothelial cells.
 Chocolate cysts: hemorrhage from interstitial capillaries.
Chocolate cysts: hemorrhage from interstitial capillaries.
 Monitor and treat amblyopia, strabismus, and anisometropia, particularly after hemorrhage.
Monitor and treat amblyopia, strabismus, and anisometropia, particularly after hemorrhage.
 Drain hemorrhage.
Drain hemorrhage.
 Avoid surgical resection because of infiltrative nature.
Avoid surgical resection because of infiltrative nature.
 Debulk lesion anteriorly as needed.
Debulk lesion anteriorly as needed.
 Orbital varices: weakened venous system that enlarges with increased venous pressure (e.g., Valsalva maneuver) and enhances on CT.
Orbital varices: weakened venous system that enlarges with increased venous pressure (e.g., Valsalva maneuver) and enhances on CT.
 Most common benign primary orbital tumor of adults, particularly middle-aged women.
Most common benign primary orbital tumor of adults, particularly middle-aged women.
 Gradual proptosis, decreased vision with optic nerve compression or induced hyperopia (Figure 7.7).
Gradual proptosis, decreased vision with optic nerve compression or induced hyperopia (Figure 7.7).
 CT: well-circumscribed mass with homogeneous enhancement.
CT: well-circumscribed mass with homogeneous enhancement.
 Uncommon tumor of middle-aged adults.
Uncommon tumor of middle-aged adults.
 Similar presentation as cavernous hemangioma.
Similar presentation as cavernous hemangioma.
 May cause conjunctival engorgement and prolapse, motility restriction.
May cause conjunctival engorgement and prolapse, motility restriction.
 CT: well-circumscribed lesion.
CT: well-circumscribed lesion.
 Complete surgical excision.
Complete surgical excision.
 May recur, undergo malignant transformation, metastasize.
May recur, undergo malignant transformation, metastasize.
 Optic nerve glioma develops during the first decade.
Optic nerve glioma develops during the first decade.
 Twenty-five to fifty percent associated with neurofibromatosis type 1.
Twenty-five to fifty percent associated with neurofibromatosis type 1.
 Painless proptosis, vision loss, afferent pupillary defect.
Painless proptosis, vision loss, afferent pupillary defect.
 Chiasmal involvement may occur.
Chiasmal involvement may occur.
 Management of optic nerve gliomas is controversial because of the variability in natural history of these tumors; although most remain stable or progress slowly, others may grow rapidly.
Management of optic nerve gliomas is controversial because of the variability in natural history of these tumors; although most remain stable or progress slowly, others may grow rapidly.
 Treatment includes observation with serial MRI, chemotherapy (first line for progressive lesions), radiation (if chemotherapy fails), or surgical excision.
Treatment includes observation with serial MRI, chemotherapy (first line for progressive lesions), radiation (if chemotherapy fails), or surgical excision.
 Radiation therapy is given for nonresectable tumors with chiasmal invasion.
Radiation therapy is given for nonresectable tumors with chiasmal invasion.
 Surgical excision involves optic nerve resection and therefore results in blindness; thus, tumor should be excised for growth toward the chiasm or for a blind proptotic eye.
Surgical excision involves optic nerve resection and therefore results in blindness; thus, tumor should be excised for growth toward the chiasm or for a blind proptotic eye.
 Intracranial meningioma:
Intracranial meningioma:
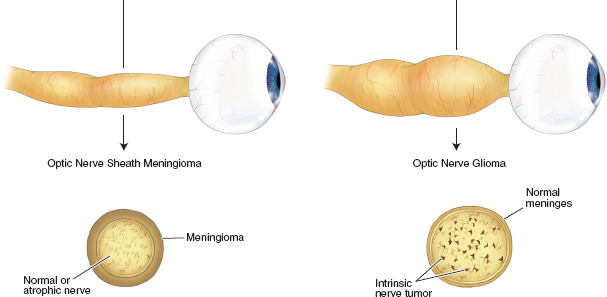
 Optic nerve meningioma (Figure 7.8):
Optic nerve meningioma (Figure 7.8):
 Meningothelial cells in various patterns.
Meningothelial cells in various patterns.
 Psammoma bodies (calcified whorls of degenerated cells).
Psammoma bodies (calcified whorls of degenerated cells).
 Meningiomas are slow-growing tumors that may be observed while following the patient carefully for vision loss, motility disturbances, or ocular exposure that is due to proptosis.
Meningiomas are slow-growing tumors that may be observed while following the patient carefully for vision loss, motility disturbances, or ocular exposure that is due to proptosis.
 Intracranial meningioma:
Intracranial meningioma:
 Optic nerve meningioma:
Optic nerve meningioma:
 Infiltrative, well-vascularized tumor within eyelid, orbit.
Infiltrative, well-vascularized tumor within eyelid, orbit.
 Upper lid lesion: “bag of worms,” S-shaped eyelid.
Upper lid lesion: “bag of worms,” S-shaped eyelid.
 Associated with neurofibromatosis type 1.
Associated with neurofibromatosis type 1.
 Higher incidence of glaucoma in the ipsilateral eye.
Higher incidence of glaucoma in the ipsilateral eye.
 Painless proptosis in middle-aged individuals.
Painless proptosis in middle-aged individuals.
 Ten percent of schwannomas occur in patients with neurofibromatosis.
Ten percent of schwannomas occur in patients with neurofibromatosis.
 Usually benign with slow progression, rare malignant transformation.
Usually benign with slow progression, rare malignant transformation.
 CT: well-circumscribed ovoid tumor.
CT: well-circumscribed ovoid tumor.
 Encapsulated proliferation of Schwann cells.
Encapsulated proliferation of Schwann cells.
 Antoni A: tightly packed Schwann cells; Verocay body (palisading nuclei).
Antoni A: tightly packed Schwann cells; Verocay body (palisading nuclei).
 Antoni B: loosely arranged Schwann cells; degeneration within the tumor.
Antoni B: loosely arranged Schwann cells; degeneration within the tumor.
 Most common metastatic orbital lesion in children.
Most common metastatic orbital lesion in children.
 Primary malignancy located in the adrenal medulla and less commonly in the cervical ganglia that can result in ipsilateral Horner syndrome.
Primary malignancy located in the adrenal medulla and less commonly in the cervical ganglia that can result in ipsilateral Horner syndrome.
 Abrupt ecchymotic proptosis, bilateral in about 50%.
Abrupt ecchymotic proptosis, bilateral in about 50%.
 Sudden onset of unilateral proptosis.
Sudden onset of unilateral proptosis.
 Eyelid swelling, injection, ecchymosis.
Eyelid swelling, injection, ecchymosis.
 Pain and decreased vision are uncommon.
Pain and decreased vision are uncommon.
 Proptosis (80% to 100%), downward and lateral displacement of globe.
Proptosis (80% to 100%), downward and lateral displacement of globe.
 Palpable mass: superonasal quadrant in 70%.
Palpable mass: superonasal quadrant in 70%.
 Ptosis: may be the first sign.
Ptosis: may be the first sign.
 Conjunctival and lid edema and erythema.
Conjunctival and lid edema and erythema.
 CT: Look for bony destruction; lesion may appear circumscribed or infiltrative.
CT: Look for bony destruction; lesion may appear circumscribed or infiltrative.
 Immediate biopsy.
Immediate biopsy.
 Metastatic workup: CXR, bone marrow biopsy, lumbar puncture.
Metastatic workup: CXR, bone marrow biopsy, lumbar puncture.
 Embryonal:
Embryonal:
 Alveolar:
Alveolar:
 Pleomorphic:
Pleomorphic:
 Botyroid embryonal:
Botyroid embryonal:
 Rapid progression of tumor with bony invasion and metastasis to lymph nodes, lung, bone marrow, and brain.
Rapid progression of tumor with bony invasion and metastasis to lymph nodes, lung, bone marrow, and brain.
 Prognosis is based on staging of disease and histopathologic features with range from best to worse prognosis: adult pleomorphic > embryonal (including botyroid) > alveolar.
Prognosis is based on staging of disease and histopathologic features with range from best to worse prognosis: adult pleomorphic > embryonal (including botyroid) > alveolar.
 When rhabdomyosarcoma is suspected, an immediate biopsy should be performed (complete or near-complete excisional biopsy), with an appropriate metastatic workup to stage the disease.
When rhabdomyosarcoma is suspected, an immediate biopsy should be performed (complete or near-complete excisional biopsy), with an appropriate metastatic workup to stage the disease.
 Radiation therapy and chemotherapy are the mainstays of treatment, resulting in a 95% 5-year survival rate.
Radiation therapy and chemotherapy are the mainstays of treatment, resulting in a 95% 5-year survival rate.
 Can involve any part of the orbit, following an indolent course.
Can involve any part of the orbit, following an indolent course.
 Middle-aged adults.
Middle-aged adults.
 Local recurrence with incomplete excision.
Local recurrence with incomplete excision.
 Spindled-shaped cells uniformly reactive to CD34, unlike other benign orbital tumors.
Spindled-shaped cells uniformly reactive to CD34, unlike other benign orbital tumors.
 Complete surgical excision.
Complete surgical excision.
 Tender palpable superotemporal mass, rhinorrhea.
Tender palpable superotemporal mass, rhinorrhea.
 First decade of life.
First decade of life.
 Results from abnormal accumulations of Langerhans cells—histiocytes normally found in the epidermis.
Results from abnormal accumulations of Langerhans cells—histiocytes normally found in the epidermis.
 Eosinophilic granuloma: solitary lytic bone lesion with soft tissue involvement.
Eosinophilic granuloma: solitary lytic bone lesion with soft tissue involvement.
 Hand-Schüller-Christian syndrome: triad of diabetes insipidus, exophthalmos, and multifocal bony lesions.
Hand-Schüller-Christian syndrome: triad of diabetes insipidus, exophthalmos, and multifocal bony lesions.
 Letterer-Siwe: fulminant fatal systemic disease with visceral involvement.
Letterer-Siwe: fulminant fatal systemic disease with visceral involvement.
 Granulomatous-histiocytic infiltrate.
Granulomatous-histiocytic infiltrate.
 Electron microscopy: tennis-racket–shaped Birbeck granules.
Electron microscopy: tennis-racket–shaped Birbeck granules.
 Progressive, painless gradual proptosis.
Progressive, painless gradual proptosis.
 Motility disturbances.
Motility disturbances.
 Firm nodular anterior orbital mass.
Firm nodular anterior orbital mass.
 Subconjunctival salmon patch.
Subconjunctival salmon patch.
 Bilateral disease in 25% of cases.
Bilateral disease in 25% of cases.
 Lacrimal gland involvement.
Lacrimal gland involvement.


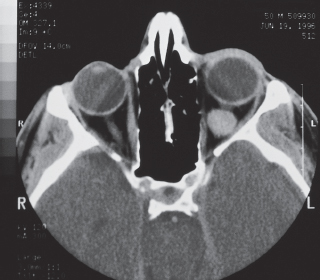
 Imaging:
Imaging: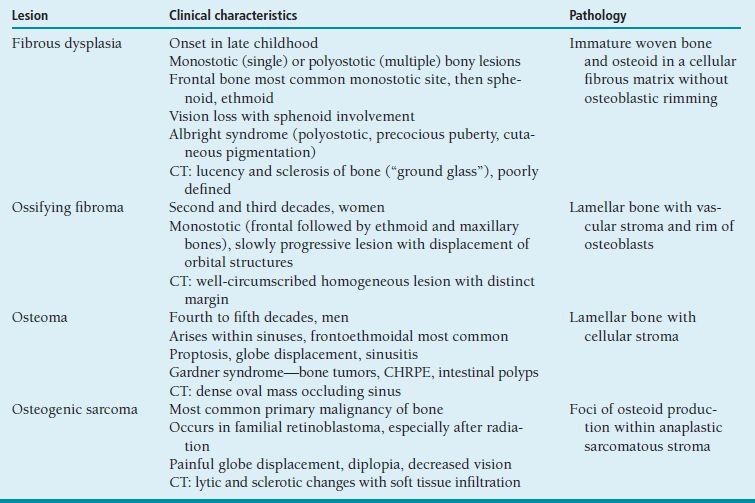
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


