Glaucomas Associated with Ocular Inflammation
Any portion of the eye can be affected by inflammatory processes, including the uveal tract (i.e., uveitis), cornea (i.e., keratitis), sclera (i.e., scleritis), and episclera (i.e., episcleritis). Uveitis is by far the most common of these diseases and can have an acute or chronic course. Most of the acute cases involve the anterior uvea (i.e., iritis or iridocyclitis), whereas the chronic forms, which have been defined as persisting for 3 months or longer, have been subclassified into four groups: anterior uveitis (i.e., iritis or iridocyclitis), intermediate uveitis (i.e., pars planitis), posterior uveitis (i.e., choroiditis or chorioretinitis), and anterior and posterior uveitis (i.e., panuveitis) (1). The relative frequencies of these four types of chronic uveitis, based on a survey of 400 consecutive patients from a uveitis service in Israel, were 45.8%, 15.3%, 14.5%, and 24.5%, respectively (1), similar to results of studies conducted in the United States and England (2–4).
The form of ocular inflammation that most frequently produces intraocular pressure (IOP) elevation is iridocyclitis (5). When glaucoma is associated with other types of ocular inflammation, there is usually secondary involvement of the anterior uveal tract. We therefore consider the clinical forms of iridocyclitis and the mechanisms and management of the associated glaucomas and then review the other forms of ocular inflammation that may be associated with glaucoma. The clinician should also keep in mind that the differential diagnosis of a patient who presents with “uveitis” and refractory glaucoma includes serious intraocular disease, such as infection (e.g., endophthalmitis), tumor (e.g., melanoma, lymphoma), acute angle-closure glaucoma, neovascular glaucoma, and secondary reaction to intraocular foreign body.
IRIDOCYCLITIS
Terminology
The general forms of iridocyclitis are classified primarily according to the clinical presentation and duration of active disease. A specific case of iridocyclitis, however, may manifest one or all of these clinical forms at different times during the course of the disease.
Acute Iridocyclitis
The characteristic history for this type of iridocyclitis is the sudden onset of mild to moderate ocular pain, photophobia, and blurred vision. Physical examination typically reveals ciliary flush, slight constriction of the pupil, and variable degrees of aqueous flare and cells (Fig. 22.1). In many cases, one will find inflammatory precipitates on the corneal endothelium (keratic precipitates). The IOP is often lower than in the fellow eye, although some patients present with a marked elevation of the pressure, which may be associated with severe pain and corneal edema.
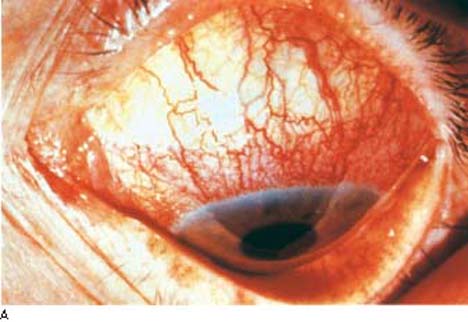
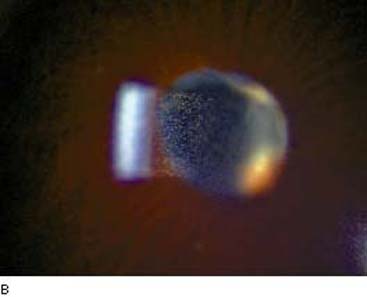
Figure 22.1 A: Slitlamp view of an eye with acute iritis shows the typical ciliary flush (arrows). (Courtesy of Gary N. Foulks, MD.) B: Slitlamp view of cell and flare in a patient with active uveitis. (Courtesy of Joseph A. Halabis, OD.)
Subacute Iridocyclitis
Some cases of ocular inflammation produce minimal or no symptoms. The diagnosis may be made during a routine eye examination or as part of a workup for a related systemic disease. This form of iridocyclitis can have serious consequences because complications such as associated glaucoma may go undetected until advanced damage has occurred.
Chronic Iridocyclitis
The clinical presentation in this form of iridocyclitis ranges from acute to subacute but is characterized by a protracted course of months to years, often with remissions and exacerbations. Complicating sequelae include the formation of posterior and peripheral anterior synechiae, cataracts, and band keratopathy. This form of iridocyclitis is particularly likely to cause glaucoma. In a study of 100 patients with uveitis, all of whom had anterior uveal involvement, glaucoma was present in 23 cases, of which 20 represented chronic uveitis and 3 acute uveitis (6).
Clinical Forms of Iridocyclitis and Glaucoma
Acute Anterior Uveitis
This is the most common form of ocular inflammation, with a lifetime cumulative incidence of approximately 0.2% in the general population (7). It represents a group of conditions characterized by acute iridocyclitis, although the pathogenesis of most cases is unknown except for the close association in many patients with the genetic marker HLA-B27. Patients with acute anterior uveitis are often classified as HLA-B27–positive and HLA-B27–negative populations. HLA-B27–positive acute anterior uveitis appears to represent a distinct clinical entity, accounting for approximately half of all cases in white patients (8). Compared with patients with HLA-B27–negative acute anterior uveitis, the HLA-B27–positive form occurs more in white individuals, has a younger age of onset (median age in the early 30s), has a slight preponderance of men, and is typically unilateral or unilateral alternating, although bilateral cases do occur (9–11). Ocular findings may be severe, such as fibrin in the anterior chamber, although mutton-fat keratic precipitates are not a typical finding (8). Ocular complications, including cataracts, posterior synechiae, elevated IOP, and cystoid macular edema, are also more common than in HLA-B27–negative cases (9–11), although the long-term visual outcome is not significantly different (9,10). Anterior segment fluorophotometry suggests that HLA-B27–positive patients have more severe inflammation on the basis of blood–aqueous barrier disruption (12).
Rheumatologic complications, including ankylosing spondylitis, are seen in one half to two thirds of HLA-B27–positive patients, but they are uncommon in the HLA-B27–negative population (8–10). The former patients also have a higher-than-normal prevalence of first-degree relatives with HLA-B27– positive acute anterior uveitis and ankylosing spondylitis (11,13).
Some topical glaucoma medications have been associated with anterior uveitis. These include metipranolol and brimonidine (14,15). Whether latanoprost is also implicated remains a subject of controversy (16).
Patients with acute anterior uveitis usually respond to nonspecific anti-inflammatory therapy (as discussed later in this chapter), although it is important to rule out related ocular or systemic disease, which may require more specific therapy (17).
Sarcoidosis
This multisystem inflammatory disorder of uncertain origin has a predilection for young adults and blacks. The typical histopathologic finding is noncaseating granulomas, and systemic involvement commonly includes pulmonary hilar lymphadenopathy, peripheral lymphadenopathy, and cutaneous lesions. In a review of 532 cases of sarcoidosis, 202 (38%) had ocular involvement (18), but in another survey of 159 patients with systemic sarcoidosis, more than one half presented with ocular lesions as the initial manifestation (19). Ocular findings include chorioretinitis, retinal periphlebitis, and occasional involvement of the optic nerve, orbit, or lacrimal glands, although the most common ocular abnormality is anterior uveitis (18,19).
Iridocyclitis. Acute iridocyclitis with the previously described features of ciliary flush, aqueous flare and cells, and occasional fine or large (mutton-fat) keratic precipitates was noted in approximately 15% of 202 patients with ocular sarcoid (18). In the acute phase, the inflammation was usually unilateral. The most common ocular manifestation of sarcoidosis is a chronic granulomatous uveitis, which was reported in more than one half of the 202 cases (18). It is more often bilateral, has a protracted course, and is typified by mutton-fat keratic precipitates, synechiae, and iris nodules (Fig. 22.2). The nodules, which were seen in 23 (11.4%) of the 202 cases of ocular sarcoid (18), may involve the pupillary border (Koeppe nodules) and stroma of the iris (Busacca nodules), as well as the anterior chamber angle and the ciliary body (19,20). In one series of 102 eyes of 52 patients with ocular sarcoidosis, 35% of eyes had iris nodules, 49% had nodules in the angle, and 42% had ciliary nodules (20). Gonioscopy may also reveal inflammatory precipitates on the trabecular meshwork and whitish spots on the ciliary body band, which hyperfluoresce during fluorescein gonioangiography and may represent granulomas of the ciliary body (21,22).
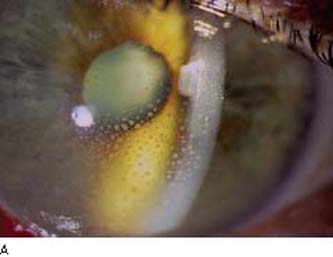
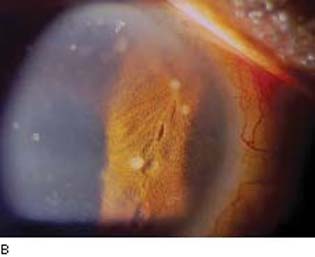
Figure 22.2 A: Slitlamp view of an eye with sarcoid uveitis shows the typical large (“mutton-fat”) keratic precipitates. B: Busacca iris nodules in a patient with sarcoid uveitis. (Courtesy of Joseph A. Halabis, OD.)
Chronic ocular sarcoidosis is associated with a worse visual prognosis than the acute form. In a series of 21 patients with sarcoid uveitis, 8 had a monophasic course and a favorable visual outcome, and 13 had a relapsing course with severe visual loss in five eyes (23). The course of the ocular disease does not always parallel that of the systemic manifestations. In one series of 33 patients with ocular and systemic sarcoidosis, all had chronic systemic manifestations, defined as a minimum duration of 5 years, whereas the anterior uveitis was chronic in only 18 patients (24). In another series of patients with chronic ocular sarcoidosis, approximately one half had no systemic manifestations (23). Sarcoidosis may also masquerade as many other conditions. In one report, five patients with clinical signs compatible with Fuchs heterochromic uveitis (discussed later) were found to have sarcoidosis on the basis of elevated serum angiotensin-converting enzyme levels or a positive Kveim test result (25).
Glaucoma. This complication of the iridocyclitis occurred in 22 of the 202 patients (10.9%) with ocular sarcoid (18). Chronic uveitis and associated glaucoma are poor prognostic signs, with 8 of 11 such patients experiencing severe visual loss in one study (24). The most common mechanism of glaucoma associated with the iridocyclitis of sarcoidosis is obstruction of the trabecular meshwork by inflammatory debris or nodules (26). A more chronic form of glaucoma may be associated with inflammatory cell infiltration around the inner and outer walls of the Schlemm canal and with iris bombé or goniosynechiae (26,27), and a subacute form has been described with precipitates on the trabecular meshwork (21). Neovascularization of the iris and angle has also been reported as a mechanism of glaucoma in association with sarcoidosis (28).
Juvenile Rheumatoid Arthritis
Juvenile rheumatoid arthritis is a spectrum of arthritic disorders in children. One form is characterized by monarticular, or pauciarticular (involvement of four joints or less) onset, a predilection for girls, and minimal additional systemic manifestations. Other types of juvenile rheumatoid arthritis have polyarticular onset or additional acute systemic involvement.
Iridocyclitis. The prevalence of iridocyclitis in patients with the monarticular or pauciarticular form of juvenile rheumatoid arthritis has been variously reported at 16%, 19%, and 29% (20,29,31), whereas the other types of juvenile rheumatoid arthritis are rarely associated with this ocular finding (30–34). Juvenile rheumatoid arthritis is by far the most common systemic finding in children who have anterior uveitis associated with a specific systemic disease, accounting for 81% of one large series (35). The ocular inflammation may have an acute onset, with the typical features of acute iridocyclitis. However, many cases are asymptomatic, emphasizing the need for periodic ocular examinations of children with juvenile rheumatoid arthritis (30,31). The onset of the arthritis typically precedes that of the uveitis, although iridocyclitis may persist in adult life, whereas the arthritis usually disappears (32). Children with iridocyclitis rarely have a positive serology for rheumatoid factor, but they frequently have antinuclear antibody and HLA-B27 antigen, and some eventually are found to have typical ankylosing spondylitis (32,34).
Complications that may cause significant visual loss in children with iridocyclitis and juvenile rheumatoid arthritis include cataracts, band keratopathy, and glaucoma. These are more common when the uveitis is the initial manifestation. In one series, 67% of such patients had a poor visual outcome, compared with only 6% of those in whom the arthritis preceded the uveitis (36). The prevalence and severity of the complications and visual loss also correlate with the degree and duration of the ocular inflammation. In one report of 60 patients, an aggressive stepladder, steroid-sparing, therapeutic algorithm (topical and regional corticosteroids, systemic nonsteroidal anti-inflammatory drugs, systemic steroids, and systemic immunosuppressive chemotherapy) was thought to control the iridocyclitis while reducing the prevalence of cataract formation and retinal pathology (37). Those cases requiring cataract surgery responded well to phacoemulsification and anterior vitrectomy after at least 3 months of complete freedom from inflammation (37).
Glaucoma. The reported prevalence of glaucoma in children with juvenile rheumatoid arthritis and iridocyclitis ranges from 14% to 27% (32–34,36). Glaucoma is a particularly serious complication, with one of the eyes in one study having a vision of 20/200 or less (34). The glaucoma mechanism is usually a pupillary block, although it may also be related to alterations in the trabecular meshwork early in the course of the disease. Histopathologic studies of two advanced cases revealed peripheral anterior synechiae and occlusion of the pupil in one (38), and a dense inflammatory infiltrate composed primarily of plasma cells in the iris and ciliary body with angle closure in the other patient (39). Treatment is typically difficult, with many eyes responding only partially to corticosteroids. The addition of nonsteroidal anti-inflammatory agents may be helpful in these cases (33,34). Antiglaucoma drugs may be required to control IOP elevation, and glaucoma surgery is occasionally needed, although reported results in these cases are poor (32).
Ankylosing Spondylitis: Marie–Strumpell Disease
Ankylosing spondylitis is a form of arthritis that typically involves the cervical or lumbosacral spine and is associated with an intermittent acute iridocyclitis in 3.5% to 12.5% of reported cases (30). A high percentage of these patients have the HLA-B27 antigen (40). Recurrent uveitis may precede the arthritic symptoms, and there is evidence, based on HLA typing and sensitive bone scans, that the ocular inflammation may occur in the absence of overt symptoms or radiologic evidence of the spondylitis (41). There is an apparent overlap between this condition and HLA-B27–positive acute anterior uveitis and the iridocyclitis with juvenile rheumatoid arthritis (8–13,34). Glaucoma may result from trabecular damage or synechiae formation.
Pars Planitis
Pars planitis is a protracted ocular inflammatory disorder that has also been referred to as intermediate uveitis or chronic cyclitis (1,42). It primarily involves the ciliary body. Typical findings include a “snowbank” appearance of the vitreous base overlying the pars plana inferiorly, retinal phlebitis, and a cystoid maculopathy (43). In a series of 100 cases with a 4- to 20-year follow-up, the incidence of glaucoma was 8% (42), whereas another group of 58 eyes had glaucoma in 7% (43). A clinicopathologic study of seven cases of pars planitis revealed glaucoma in five, and possible mechanisms of pressure elevation included peripheral anterior synechiae, iris bombé, and rubeosis iridis (44). Topical corticosteroid and antiglaucoma therapy may be effective in some cases. Decreased visual acuity, which is usually caused by cystoid maculopathy, may require the long-term use of oral and periocular steroids, cryotherapy in the area of the snowbank, and systemic antimetabolites as the final step (43).
Glaucomatocyclitic Crisis: Posner–Schlossman Syndrome
In 1948, Posner and Schlossman (45) described a uniocular disease in young to middle-aged adults, which was characterized by recurrent attacks of mild anterior uveitis with marked elevations of IOP. Many patients have associated systemic disorders, including various allergic conditions and gastrointestinal diseases, most notably peptic ulcers (46). In one series of 22 patients, HLA-Bw54 was present in 41%, suggesting that immunogenetic factors play an important role in the pathogenesis of glaucomatocyclitic crisis (47). The possible role of herpes simplex virus was also suggested by a study that revealed DNA evidence of the virus in all aqueous specimens of 3 patients during acute attacks, but in none of 10 healthy controls (48).
Iridocyclitis. The typical symptoms are slight ocular discomfort, blurred vision, and halos, which last several hours up to a few weeks or, rarely, longer and tend to recur on a monthly or yearly basis (45). Physical findings are minimal, with occasional mild ciliary flush, slight pupillary constriction, and corneal epithelial edema. Hypochromia of the iris is not a consistent finding, but it has been reported in up to 40% of various series (49). Early segmental iris ischemia with late congestion and leakage on fluorescein angiography has also been described (50). Slitlamp biomicroscopy reveals occasional faint flare and a few fine, nonpigmented keratic precipitates, and gonioscopy shows a normal, open angle with occasional debris and the characteristic absence of synechiae (45,49).
Glaucoma. The IOP is typically elevated in the range of 40 to 60 mm Hg and coincides with the duration of the uveitis. IOP and facility of aqueous outflow usually return to normal between attacks, although severe cases with optic nerve head and visual field damage have been reported (51,52). One study found that patients with 10 years or more of disease have a 2.8 times higher risk of developing glaucomatous disc and field damage, compared with patients with fewer than 10 years of disease (53). The glaucoma may be related to inflammatory changes in the trabecular meshwork. Histologic evaluation of a trabeculectomy specimen obtained during an acute attack revealed numerous mononuclear cells in the meshwork (54). Other theories of mechanism include increased aqueous production, possibly due to elevated levels of aqueous prostaglandins (55), and an association with chronic open-angle glaucoma (50,51). During attacks, most cases can be controlled with corticosteroids and antiglaucoma agents that reduce aqueous production (37,56). Apraclonidine has been reported to be especially effective during acute attacks (57). Rare, severe cases, however, may require filtering surgery (52,54).
Fuchs Heterochromic Cyclitis
In 1906, Fuchs (58) described a condition characterized by mild anterior uveitis, heterochromia, cataracts, and occasional glaucoma (Fig. 22.3A). The similarities and differences between this disease and glaucomatocyclitic crisis should be observed to avoid confusing the two. Fuchs heterochromic cyclitis (also referred to as uveitis or iridocyclitis) is usually unilateral, although bilateral involvement has been reported in up to 13% of the cases (59). The typical age of onset is in the third or fourth decade and there is an equal incidence between men and women (59,60). It is said to be the most commonly misdiagnosed form of uveitis (61), especially in black patients in whom the heterochromia may be less obvious (62).
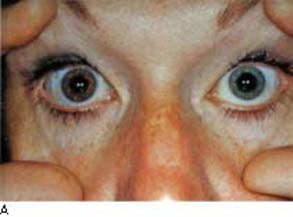
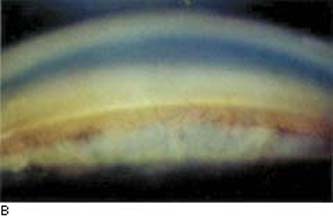
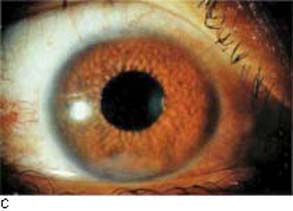
Figure 22.3 Some features of Fuchs heterochromic iridocyclitis. A: Heterochromia. B: The anterior chamber angle is open and characteristically free of synechiae, although fine vessels can be seen extending onto the trabecular meshwork. These vessels can be associated with an inflammatory membrane over the angle that may impede aqueous outflow. C: Iris nodules are seen in some patients with Fuchs heterochromic iridocyclitis.
Several etiologic theories have been considered; the most likely mechanism is true inflammation of immunologic origin, possibly related to depression of suppressor T-cell activity (61). Cellular immunity to corneal antigens has been found in most patients (63), with autoantibodies against corneal epithelium in almost 90% of cases (64). Immune deposits have been found in the vessel walls of iris biopsy specimens (65). The search for HLA-linked genetic factors is inconclusive, although preliminary evidence suggests a decrease in the frequency of HLA-CW3 (66). Fuchs heterochromic cyclitis has been reported in a father and son with associated retinitis pigmentosa (67), although the reported discordance in monozygotic twins suggests little or no genetic predisposition (68). A few patients have associated congenital Horner syndrome, suggesting the possibility of a neurogenic mechanism in these cases (69). A clinical association between Fuchs heterochromic cyclitis and toxoplasmosis raised the possibility of a causal relationship (70,71), although a study of 88 patients with the former condition revealed no association with toxoplasmosis by indirect immunofluorescence antibody tests, enzyme-linked immunosorbent assay, or cellular immunity tests to toxoplasma antigen (72). Clinical similarities with sarcoidosis have also been reported (25).
Iridocyclitis. The uveitis in this disease is mild and tends to run a single, very protracted course, although it may be intermittent initially. The patient is usually unaware of any difficulty until visual disturbance, primarily from cataract formation, becomes apparent. Although hypochromia of the iris is more common than in glaucomatocyclitic crisis, it is not a constant feature, and tends to develop gradually during the course of the disease (59,60). In one series, it was seen in 92% of 54 white patients and 76% of 13 black patients (62).
Gross signs of ocular inflammation are typically absent, although slitlamp biomicroscopy may reveal minimal aqueous flare and cells. Characteristic fine, stellate keratic precipitates are usually seen on the lower half of the cornea but may also involve the upper half (73). The iris frequently has extensive stromal atrophy, and transillumination of the iris is reported to demonstrate a characteristic light, even translucence (74). One study revealed translucency not only of the iris but also of the surrounding ocular wall (75). Electron microscopic studies of the iris have revealed a scant number of deep stromal melanocytes with immature melanin granules, abundant plasma cells, an increase in mast cells, and a membranous degeneration of nerve fibers (76,77).
Patients may have neovascularization of the anterior chamber angle and iris, and nodules on the iris (66) (Fig. 22.3B, C). The nodules typically occur along the pupillary border, similar to the Koeppe nodules of sarcoidosis, although may appear across the whole surface of the iris (78). In one series, the nodules were seen in 20% of white patients and 30% of black patients (62). The finding of unilateral iris nodules may be especially helpful in making the diagnosis of Fuchs cyclitis in black individuals, in whom the heterochromia may be less apparent (78). Anterior segment fluorescein angiography has shown delayed filling, sector ischemia, leakage, and neovascularization, and fluorophotometry has revealed an abnormal permeability of the blood–aqueous barrier (79–81). A high percentage of patients may also have chorioretinal scars, which are often consistent with toxoplasmosis (70,71,82,83).
Cataract. Cataract formation is a typical feature of Fuchs heterochromic cyclitis. In approaching cataract surgery in these patients, intensive perioperative steroid therapy is advised, and synechialysis may be required during the procedure (84). Postoperative complications of marked anterior uveitis, hyphema, IOP elevation, and cystoid macular edema are more common than in routine cataract surgery (84). However, most reports describe good visual outcomes with cataract extraction and posterior chamber intraocular lens implantation (84–86).
Glaucoma. IOP elevation is not as common as with glaucomatocyclitic crisis but may occur as a late, serious complication. The reported incidence varies from 13% to 59% (73,87–89), with the higher figures seen in series with long-term follow-up. The glaucoma typically persists after the uveitis has subsided. The anterior chamber angle is open and characteristically free of synechiae, although fine vessels, which may hemorrhage, are often seen extending onto the trabecular meshwork (87). Histopathologic examination of the anterior chamber angle structures in one case revealed rubeosis, trabeculitis, and an inflammatory membrane over the angle (90), whereas another study showed extensive atrophy of the Schlemm canal and the trabecular endothelium (91). The glaucoma typically does not respond to steroid therapy but requires standard medical or surgical management (60,73,89). In one series of 30 patients, maximum medical therapy was unsuccessful in 73%, but surgical interventions (mostly trabeculectomy, one half with 5-fluorouracil [5-FU]) were successful in 72% of the cases (89).
Behçet Disease
Behçet disease is a multisystem disease that is caused by an occlusive vasculitis. It is characterized by uveitis, aphthous lesions of the mouth, and ulcerations of the genitalia (92,93). Additional systemic findings include erythema nodosum, arthropathy, thrombophlebitis, and a necrotizing vasculitis of the central nervous system, which may be fatal. The most common ocular disorder is iridocyclitis. Behçet disease is relatively common in the Mediterranean basin and was found to be the most common associated condition with chronic uveitis in a uveitis service in Israel (1), although it is much less common in the United States and England (2–4). The iridocyclitis may be associated with a sterile hypopyon. In a study of 49 patients followed up for 10 years, 17 developed a hypopyon, which typically appears late in the course of the disease but was the initial finding in three patients (94). Posterior uveitis and necrotizing retinal vasculitis are also commonly found in this disorder (93,95). In the 10-year study, all patients developed anterior and posterior involvement within 2 years (94). The uveitis tends to occur late in the course of the disease and is eventually bilateral. The anterior uveitis may lead to glaucoma. All patients initially respond to steroid treatment, although the uveitis, in most cases, eventually requires cytotoxic–immunosuppressive agents such as chlorambucil (94). Even with this therapy, the prognosis is poor, with loss of useful visual acuity in 74% of eyes in the 10-year study (94).
Reiter Syndrome
Reiter syndrome is a multisystem disease that is characterized by conjunctivitis, urethritis, arthritis, and mucocutaneous lesions. It typically afflicts young men, with a high frequency of the HLA-B27 genotype (96). In a review of 113 patients, there were 98% with rheumatologic manifestations, 74% with genitourinary, 58% with ocular, and 42% with mucocutaneous findings (96). Conjunctivitis was seen in all patients with ocular manifestations and is characterized by a papillary reaction with a mucopurulent discharge. A nongranulomatous iridocyclitis without hypopyon was the second most common ocular manifestation, occurring in 12% of the total group, although glaucoma was only seen in 1 of the 113 patients.
Grant Syndrome: Glaucoma Associated with Precipitates on the Trabecular Meshwork
Chandler and Grant (97) described an uncommon form of open-angle glaucoma in which the only evidence of ocular inflammation was precipitates on the trabecular meshwork (Fig. 22.4). Because the condition is usually bilateral and the eyes are generally quiet, it may be mistaken for chronic open-angle glaucoma. However, careful goniosic examination reveals gray or slightly yellow precipitates on the meshwork and irregular peripheral anterior synechiae, which often attach to the trabecular precipitates (21). The cause is unknown, although some patients eventually develop sarcoidosis, rheumatoid arthritis, ankylosing spondylitis, episcleritis, glaucomatocyclitic crisis, or chronic uveitis (21). The glaucoma, which is presumed to be caused by inflammatory changes in the trabecular meshwork, usually clears promptly with topical corticosteroid therapy, although antiglaucoma drugs that reduce aqueous production may be temporarily required for pressure control. The condition often recurs, and the patients must be followed closely. Untreated cases may progress to synechial closure of the angle.
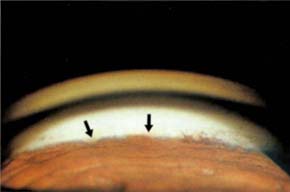
Figure 22.4 Keratitic precipitates on the trabecular meshwork (i.e., Grant syndrome).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


