Childhood Glaucomas: Clinical Presentation
Childhood glaucomas constitute a heterogenous group of disorders affecting the pediatric age-group. The previous chapter (Chapter 13) considered the general classification of these diseases, and provided a general approach to the infant or child with glaucoma. In this chapter, we highlight important features of the more common types of primary, and a few of the secondary, pediatric glaucomas, with attention to those features specific to children (since some of the secondary glaucomas also affect adults) (Table 13.1). Recall that the primary pediatric glaucomas can be broadly divided into (a) those with an isolated aqueous outflow abnormality and (b) those with associated ocular abnormalities, systemic abnormalities, or both. The former group can be further divided into (a) those presenting in the first 3 years of life (primary congenital/infantile glaucoma [PCG]) and (b) those presenting after that period but before adulthood (juvenile open-angle glaucoma [JOAG]). The latter group, often termed developmental glaucomas, comprises many different disorders, a few of which will be included in more detail in this chapter.
The secondary pediatric glaucomas are caused by a preceding process in the eye, and many are common to both adults and children, although some seem specific to children (such as glaucoma after removal of congenital cataracts). In addition, the mechanism of glaucoma in selected developmental cases (see Table 13.1) may be secondary rather than strictly primary.
PRIMARY CONGENITAL GLAUCOMAS
Classification
When they occur without a consistent association with other ocular or systemic anomalies (in other words, they seem primary), congenital glaucomas have traditionally been called PCG or primary congenital open-angle glaucoma (1). In this chapter, we will refrain from referring to other birth- or infancy-onset childhood glaucomas as PCG unless the outflow pathway defect and resultant elevated intraocular pressure (IOP) occur in apparent isolation. Newborn glaucoma, the most severe form of PCG, is apparent at birth, whereas infantile glaucoma refers to cases of PCG with clinical onset after birth but in the first 3 years of life (2). In general, the terms PCG and primary infantile glaucoma may be used interchangeably. Although PCG has also been called buphthalmos (i.e., cow’s eye) or hydrophthalmia, referring to the enlargement of the eye that may occur with this condition (3), these terms should not be used as synonyms for PCG because enlargement of the globe is seen with other childhood glaucomas if they occur early enough in life.
Primary congenital glaucoma has also been referred to as isolated trabeculodysgenesis or goniodysgenesis, to indicate that the iris and cornea are morphologically normal. Newborn glaucoma, a severe variant of PCG present at birth, is also considered by some as iridotrabeculodysgenesis (Table 13.1). PCG can therefore be considered as one form of anterior segment dysgenesis.
When primary glaucoma appears later in childhood or early adulthood, it is sometimes referred to as juvenile glaucoma (also JOAG) (4). Three years of age is generally taken as the division between PCG and JOAG, because it is at approximately this age that the eye no longer expands in response to elevated IOP (1,4). Others prefer a broader definition for juvenile glaucoma that includes all forms of open-angle glaucoma diagnosed between the ages of 10 and 35 years (5) (see JOAG, Chapter 11).
General Features
Demographic Features
The most common of the primary pediatric glaucomas, PCG has an estimated incidence of 1 in 10,000 to 20,000 live births in Western countries, while it presents more frequently in the Middle East and among the Roma population of Slovakia, where parental consanguinity may play a role in the increased incidence (6). Lacking clear sex or racial–ethnic predilection (except where consanguinity or small population may play a role), most PCG cases (65% to 80%) are bilateral, and greater than 75% present in the first year of life. About 25% of patients with PCG present initially as newborns, and more than 60% of PCG diagnoses are made in infants younger than 6 months of age (7). Nonetheless, this condition occurs much less frequently than the open-angle and angle-closure glaucomas seen in adults, and it has been estimated that the average ophthalmic practice encounters one new case of congenital glaucoma every 5 years.
Heredity
PCG occurs in both sporadic and familial patterns. Inheritance is usually autosomal recessive in familial cases, and hence, there is increased incidence with consanguinity. Three genetic loci—GLC3A, GLC3B, and GLC3C (Table 8.1)—have been identified by linkage analysis in large pedigrees with multiple affected individuals (8,9). The presence of additional loci has also been suggested (10). Thus far, two main causative genes have been reported—the CYP1B1 gene, on the GLC3A locus, and the LTBP2 gene, possibly on the GLC3C locus (9–12). The MYOC gene has also been implicated in rare cases of PCG (13–15).
The CYP1B1 gene (Online Mendelian Inheritance in Man [OMIM] number, 601771) was the first reported PCG-causing gene (9,16). It is located on chromosome 2p22–p21 on the GLC3A locus. It belongs to the cytochrome P450 superfamily of enzymes and oxidizes several compounds important to eye structure and function, including steroids, retinoids, arachidonate, and melatonin (17–22). The CYP1B1 enzyme is thought to participate in the metabolism of an unknown molecule that is important to eye development and therefore plays an important role in the development of PCG. Studies have demonstrated its expression in fetal and adult ciliary body and neuroepithelium (23). Since the CYP1B1 gene’s discovery, many PCG cohorts have been screened for CYP1B1 sequence variants. Several CYP1B1 sequence variants have been determined to cause PCG (24). The proportion of PCG patients whose disease is due to CYP1B1 mutations varies with ethnicity, ranging from 100% in the Roma population of Slovakia to 20% in Japan (25,26).
The MYOC gene (OMIM 601652) has been associated with juvenile and adult forms of open-angle glaucoma and a few rare cases of PCG (13,15,16). It is located on chromosome 1q24.3–q25.2 (27). MYOC or myocilin is also known as trabecular meshwork–induced glucocorticoid-response protein (TIGR). As its name implies, treatment of trabecular meshwork cells with glucocorticoids results in the induction of MYOC (28). It is speculated that MYOC obstructs trabecular meshwork outflow and thus causes increased IOP (27). Increased IOP may also be caused by changes in the ciliary body secondary to MYOC. Studies have revealed expression of MYOC in both the trabecular meshwork and ciliary body (29). Disease-causing MYOC sequence variants, both with and without CYP1B1 alterations, have been reported in families with early-onset open-angle glaucoma, including PCG (13–15).
LTBP2 (OMIM 602091) is the most recent gene to be associated with PCG (11,12,16). LTBP2, or latent transforming growth factor beta binding protein 2, is located on chromosome 14q24 (30). Its location is 1.5 Mb from the GLC3C locus. Whether LTBP2 and GLC3C represent the same genetic component remains to be determined. In nonocular tissues, LTBP2 functions in tissue repair and cell adhesion (31–34). The role LTBP2 plays in PCG is still unknown. Ocular expression of LTBP2 has been demonstrated in the trabecular meshwork and ciliary processes (35). Null mutations of LTBP2 have been found in consanguineous Pakistani and Iranian families, as well as Slovakian Roma, with PCG (11,12).
All siblings of any child with PCG (or early-onset JOAG) should be examined carefully; infants should be followed up closely, especially during the first year of life, to exclude this disease. A discussion of the risk of having additional children with PCG should be carried out with parents, either with the ophthalmologist or a genetic counselor.
Clinical Features
PCG is bilateral in 65% to 80% of cases (3,36), although a significant IOP elevation may occur in only one eye in 25% to 30% of the cases. Several ocular features, with the possible exception of gonioscopic findings, are not unique to the PCG, but they may be a part of any childhood glaucoma during the first few years of life. The neonatal globe is distensible and often greatly enlarges with exposure to elevated IOP. Stretching of the infant eye is not limited to the cornea and may involve the anterior chamber angle structures, sclera, optic nerve, scleral canal, and lamina cribrosa (7) (see also Chapter 13).
History
Infants with PCG usually present for ophthalmologic evaluation because the pediatrician or the parents have noticed something unusual about the appearance of the patient’s eyes or behavior. Often, corneal opacification and enlargement (resulting from elevated IOP) are the signs that signal glaucoma in the infant (Figs. 14.1, 13.2, and 14.2). In other cases, the child’s glaucoma manifests as one or more of the classic triad of findings, any one of which should arouse suspicion of glaucoma in an infant or young child: (a) epiphora (i.e., excessive tearing); (b) photophobia (i.e., hypersensitivity to light), which results from corneal edema and is manifested by the child hiding his or her face in bright lighting or even in ordinary lighting in severe cases; and (c) blepharospasm (i.e., squeezing the eyelids), which may be another manifestation of photophobia.
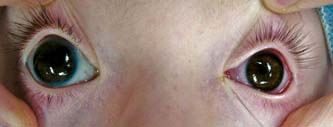
Figure 14.1 Infantile glaucoma, with asymmetric involvement. Note marked enlargement of the right, compared with the left, cornea. The IOP was higher in the right eye, where multiple Haab striae and corneal edema were present.
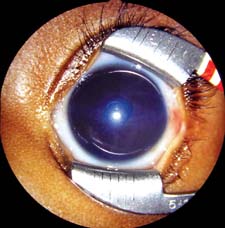
Figure 14.2 Corneal edema with a central Haab striae in the eye of a 5-month-old infant girl with PCG. IOP was 28 mm Hg on maximal tolerated medications at examination under anesthesia, several weeks after trabeculotomy was performed from the temporal limbus. Further angle surgery was planned. (See also Fig. 13.2.)
The severity of presenting signs and symptoms varies among infants with PCG, probably because of differences in the magnitude and duration of the IOP elevation. For example, newborn infants presenting with enlarged, very cloudy corneas presumably had elevated IOP in utero, whereas those with milder signs and symptoms might have experienced the IOP elevation beginning sometime after birth. Parents and health-care providers have occasionally failed to recognize glaucoma in infants with clear but enlarged corneas (37) (Fig. 14.3). Some bilateral cases may manifest with such asymmetric signs and symptoms that glaucoma is initially suspected only in the more severely affected eye. In children with glaucoma onset after 1 year of age, fewer overt signs and symptoms may occur because of the decreased expansibility of the eye.
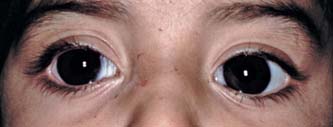
Figure 14.3 A child whose diagnosis of congenital glaucoma was delayed until 2.5 years of age. Corneal diameters were 15 mm OU. The IOP was 40 mm Hg, and the optic nerves showed total cupping; best vision was less than 20/400 OU. IOP control was achieved by using goniotomy and medications for 6 years; the patient then required trabeculectomy with use of mitomycin C for glaucoma control.
External Examination
The infant with PCG presents as an otherwise healthy child, without any systemic or facial features to suggest a different diagnosis. Often the examiner notes that the child is unusually photophobic and fussy, and parents frequently relate a history of eye rubbing.
Corneal Features
Corneal Diameter
The healthy newborn’s cornea has a horizontal diameter ranging from 9.5 to 10.5 mm, which enlarges about 0.5 to 1.0 mm in the first year of life (38–40) (Table 13.2). Distention of the globe in response to elevated IOP (buphthalmos) leads to additional enlargement of the cornea, especially at the corneoscleral junction. A corneal diameter larger than 12 mm in the first year of life is a highly suspect finding. Asymmetry in diameter between the two corneas or a corneal diameter of 13 mm or more at any age strongly suggests abnormality (7). Corneal enlargement is more obvious in asymmetric cases (Fig. 14.1). In one study, corneal diameter was found to be a more reliable guide than axial length in the assessment of congenital glaucoma (38).
Corneal Edema
Initially, corneal edema may be a direct result of the elevated IOP, producing a corneal haze that clears with normalization of the pressure. Often, there are underlying breaks in the Descemet membrane (Haab striae) that occur as the cornea stretches because of elevated IOP. These often appear acutely as areas of increased corneal edema and clouding; clinical onset may take only a matter of hours (7) (see Slitlamp section). In more advanced cases, a dense opacification of the corneal stroma may persist despite reduction of the IOP (Fig. 14.4). Results of one study suggest that the latter may result from reduced aqueous production with poor corneal nutrition (41).
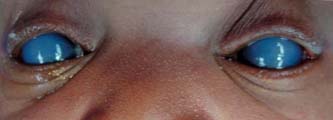
Figure 14.4 Newborn-onset congenital glaucoma with severe bilateral corneal edema and opacification. Despite IOP reduction after surgery, central corneal opacification did not clear completely.
Refractive Error
The enlargement of the globe with elevated IOP in the first 3 years of life creates a myopic shift in the refractive error, which may lead to amblyopia if significant anisometropia is present. The presence of Haab striae often produces significant astigmatism, which also contributes to amblyopia, especially in unilateral or asymmetric cases. Children between 3 and 10 years of age with elevated IOP may develop progressive myopia and astigmatism, despite a stable corneal diameter. These refractive changes have been attributed to continued scleral stretching (7).
Tonometry
Measurement of the IOP in an infant or child suspected of having PCG should ideally be performed in the office, with the child as calm as possible. Useful handheld devices include the Perkins, Tono-Pen, and ICare tonometers, while the cooperative patient older than about 3 years (and without nystagmus) can often sit for Goldmann applanation. It is important to avoid traumatizing a child to obtain the IOP, because tonometry performed in a struggling child will invariably produce falsely elevated readings, which will be useless in diagnosing PCG or assessing control of known PCG. Infants with PCG commonly present with unanesthetized IOPs in the range of 30 to 40 mm Hg, although occasionally values above or below this range occur (42). Target pressures for children with PCG depend entirely on the details of the particular case; while IOPs in the low 20-mm Hg range may be adequate for a child with healthy optic nerves and stable refraction, others with more severe disease may progress at these same IOPs and require lower target IOP (see also Chapters 13 and 40).
Measuring the IOP under anesthesia is sometimes necessary, but should be coupled with an assessment of the overall status of the eye (or eyes), together with subsequent surgical intervention when necessary. When the IOP in infants and young children is measured during general anesthesia, the possible influence of the anesthesia on IOP must be considered (see Table 13.4), with the IOP measurement taken as soon as the airway is secure. A pressure of 20 mm Hg or greater should arouse suspicion (43). In cases of unilateral PCG, asymmetry of IOP measured under anesthesia may be very helpful, even if the true IOP has been altered in this setting.
Slitlamp Examination
This portion of the examination is best performed with a portable slitlamp, with or without general anesthesia. Tears in the Descemet membrane (i.e., Haab striae) are classic findings of PCG; they may be single or multiple, and are characteristically oriented horizontally or concentric to the limbus (Fig. 14.5). They are typically associated with corneal edema in the early phases of glaucoma. Haab striae are found in about 25% of eyes with a diagnosis of PCG at birth and in more than 60% of those with that diagnosis at 6 months of age (44). These Haab striae remain as a testament to the early onset of the IOP elevation, even in late-diagnosed cases, or those rare cases with spontaneous resolution of the IOP elevation. As the IOP is normalized and the tears are repaired by endothelial overgrowth, the corneal edema may clear, but the linear opacities persist. Specular microscopy has shown that these patients also have a significantly reduced corneal endothelial cell count.

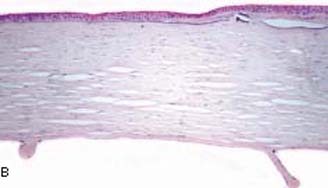
Figure 14.5 A: Haab striae in the peripheral cornea of a 10- year-old child with congenital glaucoma. IOP was controlled with angle surgery and medications, but the scar remains. (See also Figs. 13.5 and 13.6.) B: Healed breaks in Descemet membrane (Haab striae) are seen in the cornea with congenital glaucoma. The Bowman layer contains basophilic deposits (band keratopathy) as a degenerative change (stain, hematoxylin–eosin). (From Milman T. Congenital anomalies. In: Tasman W, Jaeger EA, eds. Duane’s Foundations of Clinical Ophthalmology. Vol. 3. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:chap 2.)
The anterior chamber is characteristically deep, especially when the globe is distended. The iris is typically normal, although it may have stromal hypoplasia with loss of the crypts.
Gonioscopy
Evaluation of the anterior chamber angle is essential for the accurate diagnosis of PCG. (The instruments and techniques of gonioscopy are discussed in Chapter 3.) In performing gonioscopy on infants and children under anesthesia, an infant Koeppe goniolens is recommended, together with a portable slitlamp for illumination and magnification. (See Chapter 13 for description of normal childhood gonioscopy findings.)
The anterior chamber angle has a characteristic, although slightly variable, appearance in PCG (Fig. 14.6). Usually, the iris has an insertion more anterior than that of the healthy infant, with altered translucency of the angle face rendering rather indistinct ciliary body band, trabecular mesh, and scleral spur. This translucent tissue has historically been referred to as Barkan membrane (45,46). The scalloped border of the iris pigment epithelium and the trabecular meshwork itself, often prominent in PCG, may appear through the translucent peripheral iris stroma as if viewed through a morning mist.
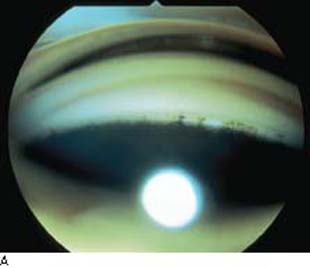
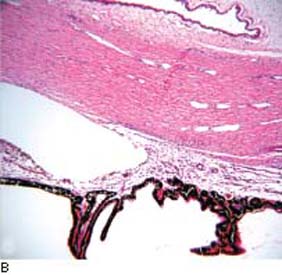
Figure 14.6 A: Gonioscopic appearance of infantile glaucoma. Note the relatively high iris insertion, with indistinct angle landmarks and fine iris processes. The angle appears wider on the right side of the photograph, at the site of prior goniotomy surgery. B: Fetal angle manifests anterior insertion of the iris root and anterior displacement of the ciliary processes. The scleral spur is poorly developed. Trabecular meshwork and Schlemm canal are poorly defined; mesenchymal tissue is present in the anterior chamber angle. (From Milman T. Congenital anomalies. In: Tasman W, Jaeger EA, eds. Duane’s Foundations of Clinical Ophthalmology. Vol. 3. Philadelphia, PA: Lippincott Williams & Wilkins; 2008:chap 2.)
Although the angle is usually avascular, loops of vessels from the major arterial circle may be seen above the iris root, which has been called the Loch Ness monster phenomenon (45). The clinical features of PCG seem to merge with other forms of developmental glaucoma. A gonioscopic assessment of more than 100 eyes with developmental glaucoma revealed a spectrum ranging from the common form described earlier, through a more cicatrized, vascularized condition, to the gross anomalies of the Axenfeld–Rieger syndrome (47).
Funduscopy
Evaluation of the optic nerve head is one of the most important methods for diagnosing PCG and for assessing the response to therapy. This is usually done with the child anesthetized or sedated, often with an undilated pupil, in which case visualization of the disc may be facilitated by using a direct ophthalmoscope with a Koeppe gonioscopy lens on the cornea or a lens designed for vitrectomy surgery (48).
Cupping of the optic nerve head proceeds more rapidly in infants than in adults and is more likely to be reversible if the pressure is lowered early enough (49–56).
Significant optic nerve cup size and asymmetry of cupping between fellow eyes suggest, but do not confirm, glaucoma in an infant. The cup-to-disc ratio exceeded 0.3 in 68% of 126 eyes with PCG examined by Shaffer and Hetherington (53), but did so in only 2.6% of 936 healthy newborn eyes examined by Richardson (57). Marked optic cup asymmetry was observed in only 0.6% of healthy eyes in the latter study, in contrast to 89% for infants with monocular glaucoma.
Visual Fields
When tested after the child becomes old enough for a reliable study (typically about 8 to 9 years of age for a child without cognitive impairment and nystagmus), the visual fields are similar to those in adult-onset glaucoma, with an initial predilection for loss in the arcuate areas (56).
Visual Acuity
Good vision may be achieved if the IOP is controlled before optic atrophy occurs. Occasionally, however, the acuity is poor despite adequate pressure control. In some cases, this is caused by optic nerve damage, corneal opacity from breaks in the Descemet membrane or persistent stromal haze, or irregular astigmatism (44,58). Other children may have normal-appearing optic nerve heads and clear media but develop amblyopia from anisometropia or strabismus (59). Retinal detachment is also an occasional cause of poor visual results (60).
Ultrasonography
Ultrasonography may be helpful in documenting progression of infantile glaucoma by recording changes in the axial length of the globe (40,61,62). It has also been reported that the axial length may decrease by as much as 0.8 mm after surgical reduction of the IOP (62). This change in axial length may be evident within days after a significant IOP reduction, especially in aphakic eyes of infants after filtration surgery or implantation of a glaucoma drainage device (Freedman SF, personal experience). Ultrasonography may also be helpful when glaucoma drainage-device surgery is being contemplated, because the size of the proposed implant reservoir may be limited by the globe size (see Chapter 40). After such surgery, ultrasonography can be helpful in confirming the presence of fluid around the device’s reservoir, especially in patients in whom the bleb cannot easily be visualized in the office setting (Fig. 14.7).
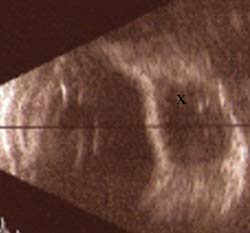
Figure 14.7 In an eye with a functional Ahmed glaucoma drainage device, B-scan ultrasonography reveals fluid-filled space surrounding the implant reservoir in the superotemporal quadrant (indicated by an x), indenting the sclera. In the office setting, the bleb was difficult to visualize but its presence was confirmed easily by using ultrasonography.
Other Testing Techniques
Corneal pachymetry to measure central corneal thickness may prove useful after corneal edema has cleared, to help set a target IOP for a particular eye. Children with PCG generally have a relatively low central corneal thickness, presumably because of the enlargement of their corneas in early infancy (63) (see Chapter 13). Care should be taken not to “adjust” the measured IOP based on central corneal thickness readings, but rather to guide the determination of the eye’s target IOP.
Other technologies, such as optical coherence tomography (OCT) (64) (Chapter 4), may prove useful in assessing nerve fiber layer loss in children too young to perform reliable visual field testing.
Etiology
Normal Development of the Anterior Ocular Segment
A basic understanding of the normal development of the anterior ocular segment is necessary before considering the theories of mechanism for congenital glaucoma or for any of the developmental glaucomas with associated anomalies.
General Development
The lens vesicle begins to develop as an invagination of surface ectoderm during the third week of gestation and separates from the latter structure by the sixth week (65). A study of 53 human embryos showed that the adhesion between the lens vesicle and presumptive corneal epithelium at the 8-mm stage is replaced by a “clear zone” at the 12.5-mm stage (66). The same study suggested that the formation of the eye is influenced by signals from neural and pigmented layers and that the lens, with its relatively large size and high mitosis, participates in the early embryogenesis of the rudimentary anterior chamber.
At the same time that the lens vesicle is separating from surface ectoderm, the optic cup, which arises from neural ectoderm, has reached the periphery of the lens, and a triangular mass of undifferentiated cells overrides the rim of the cup and surrounds the anterior periphery of the lens. From this tissue mass will arise portions of the cornea, iris, and the anterior chamber angle structures.
Neural Crest Cell Contribution
The undifferentiated cell mass destined to become the cornea, iris, and anterior chamber angle was originally thought to be derived from mesoderm. Subsequent studies, however, indicated that the tissue is of cranial neural crest cell origin. Johnston and colleagues (67) studied orofacial development in chick embryos. By using these models, it was determined that corneal endothelium and the stroma, iris, ciliary body, and sclera are of neural crest origin, except for the associated vascular endothelium, which is derived from mesodermal mesenchyme. Immunohistochemical studies have provided support for the concept that cells of human trabecular meshwork are also of neural crest origin by showing evidence of neuronal cell–specific enolase (68,69). These cells were found in the anterior region of the meshwork and in the inner uveal beams (68,69), whereas the cells lining the Schlemm canal were found to share many immunophenotypical features with vascular endothelial cells (69).
Development of Cornea and Iris
From the mass of undifferentiated cells, three waves of tissue come forward between the surface ectoderm and lens. The first of these layers differentiates into the primordial corneal endothelium by the eighth week and subsequently produces Descemet membrane, and the second wave grows between the corneal endothelium and epithelium to produce the stroma of the cornea (70,71). The third wave insinuates between the primordia of the cornea and the lens and gives rise to the pupillary membrane and the stroma of the iris. In later months, the pigment epithelial layer of the iris develops from neural ectoderm.
Development of Anterior Chamber Angle
The aqueous outflow structures in the anterior chamber angle appear to arise from the mesenchymal mass of neural crest cell origin. The precise details of this development are not fully understood. Theories have included atrophy or resorption (i.e., progressive disappearance of portions of fetal tissue), cleavage (i.e., separation of two pre-existing tissue layers due to differential growth rates), and rarefaction (i.e., mechanical distention due to growth of the anterior ocular segment) (46,65,72,73). Subsequent work suggests that none of these concepts is completely correct.
Anderson (74) studied 40 healthy fetal and infant eyes by light and electron microscopy and found that the anterior surface of the iris at 5 months’ gestation inserts at the edge of the corneal endothelium, covering the cells that are destined to become trabecular meshwork. This appears to be what Worst (45) called the fetal pectinate ligament, separating the corneoscleral meshwork primordium from the anterior chamber angle. Anderson observed a posterior repositioning of the anterior uveal structures in relation to the cornea and sclera in progressively older tissue specimens, presumably because of the differential growth rates. At birth, the insertion of the iris and ciliary body is near the level of the scleral spur, and the posterior migration of these structures continues for about the first year of life.
There is some difference of interpretation regarding the innermost layer of the trabecular meshwork primordium, as it is uncovered by the posteriorly receding iris. Anderson (74) thought that the smooth surface represents multilayered mesenchymal tissue, which begins to cavitate by the seventh fetal month. Others have suggested that a true endothelial layer covers the meshwork during gestation (45,75). Hansson and Jerndal (75) observed that the anterior chamber angle portion of the endothelial layer begins to flatten, with loss of clear-cut cell borders, by the seventh fetal month. During the final weeks of gestation and the first weeks after birth, the endothelial layer undergoes fenestration with migration of cells into the underlying uveal meshwork. Van Buskirk (76) observed a similar endothelial layer and its progressive fenestration in macaque monkey eyes. He noticed that fenestration and gradual retraction of this tissue occurred in the third trimester and progressed in a posterior-to-anterior direction. McMenamin (77), however, in a scanning electron microscopic study of 32 human fetal eyes, found that the endothelial layer in the iridocorneal angle was perforated by discrete intercellular gaps by 12 to 14 weeks and that the gaps between the inner uveal trabecular endothelial cells were sufficiently developed by 18 to 20 weeks to allow a route of communication between the fetal anterior chamber and primitive trabecular tissue.
McMenamin (78) also showed, in light and electron microscopic studies of human fetal eyes between 12 and 22 weeks’ gestation, that the trabecular anlage doubles in cross-sectional area, cell density decreases but the absolute number of cells increases twofold to threefold, extracellular matrix increases in a predictable fashion by 360%, and the intertrabecular spaces increase in a more variable manner by 200%. It appears that the trabecular meshwork develops by a simple process of growth and differentiation. These observations have been combined in a concept of anterior chamber angle development (79), which is depicted in Figure 14.8.
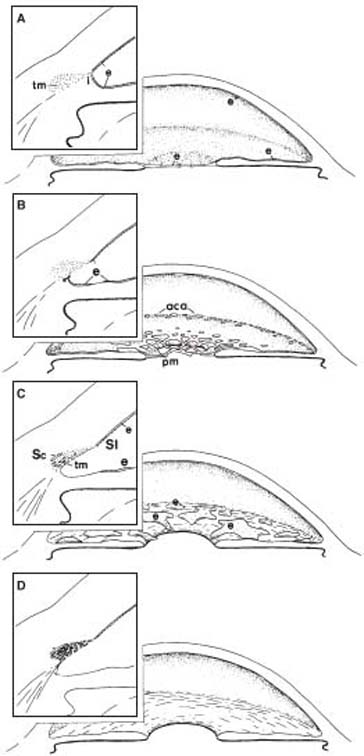
Figure 14.8 A concept of anterior chamber angle development (insets show cross-sectional views of the chamber angle). A: At 5 months’ gestation, a continuous layer of endothelium (e) creates a closed cavity of the anterior chamber (according to most studies), and the anterior surface of the iris (i ) inserts in front of the primordial trabecular meshwork (tm). B: In the third trimester, the endothelial layer progressively disappears from the pupillary membrane (pm) and iris and cavitates over the anterior chamber angle (aca), possibly becoming incorporated in the trabecular meshwork. At the same time, the peripheral uveal tissue begins to slide posteriorly in relation to the chamber angle structures (arrow). C: Development of the trabecular lamellae and intertrabecular spaces begins in the inner, posterior aspect of the primordial tissue and progresses toward the Schlemm canal (Sc) and Schwalbe line (Sl ). D: The normal anterior chamber angle is not fully developed until 1 year of age. (From Shields MB. Axenfeld–Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983;81:736, Republished with permission of the American Ophthalmological Society.)
Theories of Abnormal Development in Congenital Glaucoma
Although it is generally agreed that the IOP elevation in congenital glaucoma is caused by an abnormal development of the anterior chamber angle that leads to obstruction of aqueous outflow, there is no universal agreement on the nature of the developmental alteration. Theories of pathogenesis parallel the basic concepts regarding the normal development of the anterior chamber angle, most of which are no longer accepted as being entirely correct. We first review the major theories that have been proposed in the past and then consider how they fit with our current understanding of the developmental abnormality of congenital glaucoma.
In 1928, Mann (80) postulated that incomplete atrophy of anterior chamber mesoderm resulted in retention of abnormal tissue that blocked aqueous outflow. In 1955, Barkan (46) suggested that incomplete resorption of the mesodermal cells by adjacent tissue led to the formation of a membrane across the anterior chamber angle. This structure became known as the Barkan membrane, although its existence has not been proved histologically. Electron microscopic studies by Anderson (46,81) revealed no membrane, despite the appearance of such a structure by gonioscopy and the dissecting microscope. In 1955, Allen and colleagues (72) postulated that incomplete cleavage of mesoderm in the anterior chamber angle resulted in the congenital defect. In 1966, Worst (45) proposed a combined theory that included elements of the atrophy and resorption concepts but rejected the cleavage theory. However, all of the theories for normal development of the anterior chamber angle, on which each of the previous theories of pathogenesis was based, are no longer thought to be correct.
In 1959, Maumenee (82,83) observed an abnormal anterior insertion of the ciliary musculature into the trabecular meshwork and reasoned that this might compress the scleral spur forward and externally, narrowing the Schlemm canal. Anderson (74) and others (84) provided further histopathologic support for the high insertion of the anterior uvea into the trabecular meshwork, suggesting that it is caused by a developmental arrest in the normal migration of the uvea across the meshwork in the third trimester of gestation. Maumenee (83) also noticed the absence of Schlemm canal in some histopathologic specimens and suggested that this might be a cause of aqueous outflow obstruction in congenital glaucoma, although Anderson (81) thought it might be a secondary change.
In 1971, Smelser and Ozanics (73) explained congenital glaucoma as a failure of anterior chamber angle anlage to become properly rearranged into the normal trabecular meshwork. Subsequent light and electron microscopic studies favored this theory by showing structural changes of the uveal meshwork and, in some cases of infantile and juvenile glaucoma, a thick layer of amorphous material beneath the internal endothelium of the Schlemm canal (81,85–89). Kupfer and colleagues (90,91) emphasized the contribution of the cranial neural crest cells in the development of the anterior chamber angle and suggested that abnormal development of structures derived from these cells may result in the defects of the various forms of congenital glaucoma.
In summary, most forms of congenital glaucoma appear to result from a developmental arrest of anterior chamber angle tissue derived from neural crest cells, leading to aqueous outflow obstruction by one or more of several mechanisms. The high insertion of ciliary body and iris into the posterior portion of the trabecular meshwork may compress the trabecular beams. There may be primary developmental defects at various levels of the meshwork and, in some cases, the Schlemm canal. However, a true membrane over the meshwork does not appear to be a feature of this disorder.
Differential Diagnosis
Some of the clinical features of PCG are also found in other conditions, and these must be considered in the differential diagnosis (Table 13.2).
Excessive Tearing
In the infant, excessive tearing is most commonly caused by obstruction of the lacrimal drainage system. The epiphora of nasolacrimal duct obstruction is distinguished from that of PCG (or any infantile-onset glaucoma) in that the former condition may be associated with fullness of the lacrimal sac and often has a purulent discharge. The epiphora of PCG (and any infantile glaucoma) is frequently associated with photophobia and blepharospasm, although these three findings can also result from various external ocular disorders. Any of the several types of conjunctivitis in the infant may manifest with epiphora and a “red eye,” but photophobia is usually absent. When epiphora, photophobia, or blepharospasm accompanies a red eye, ocular inflammation (i.e., uveitis) and corneal injury or keratitis (e.g., abrasion, herpetic dendrite) should be considered.
Corneal Disorders
Large Corneas
Large corneas may represent congenital megalocornea without glaucoma or an enlarged globe due to high myopia. However, PCG (and any infantile-onset glaucoma) also typically causes progressive myopia resulting from enlargement of the globe. Infants with megalocornea often present with symmetrically enlarged, clear corneas with diameters larger than 14 mm, with deep anterior chambers, and with iridodonesis, but without elevated IOP or optic nerve cupping. Megalocornea is a rare, X-linked recessive disorder; families have been described in which some individuals have megalocornea alone, whereas others present with primary infantile glaucoma (58,92). A pedigree has been described with autosomal dominant megalocornea and congenital glaucoma in which the inheritance pattern is thought to represent germ-line mosaicism (93).
Eyes with axial myopia often show enlargement of the globe and cornea, but without elevated IOP; posterior segment examination usually demonstrates an oblique optic nerve head insertion and scleral crescent, often with suggestive chorioretinal findings. Any infant with corneal enlargement should be followed up over time for the development of elevated IOP.
Tears in Descemet Membrane
Tears in the Descemet membrane may result from forceps injury during birth (94). These tears are usually vertical or oblique, in contrast to those of congenital glaucoma (i.e., Haab striae) (Fig. 13.7), which tend to be horizontal or concentric with the limbus. Tears in the Descemet membrane may also be confused with band-like structures in posterior polymorphous dystrophy and posterior corneal vesicles (95,96). Haab striae may be distinguished from these disorders by thin, smooth areas between thickened, curled edges, contrasting with the central thickening in posterior polymorphous dystrophy and posterior corneal vesicles (95).
Corneal Opacification
Corneal opacification in infancy may be associated with various disorders (97): developmental anomalies (i.e., Peters anomaly and sclerocornea), dystrophies (i.e., congenital hereditary corneal dystrophy and posterior polymorphous dystrophy), choristomas (i.e., dermoid and dermis-like choristoma), edema due to birth trauma, intrauterine inflammation or keratitis (i.e., congenital syphilis, rubella, and herpetic infection), and inborn errors of metabolism (i.e., mucopolysaccharidoses [MPS] and cystinosis).
Other Glaucomas of Childhood
The differential diagnosis of PCG should include developmental glaucomas with associated anomalies and the childhood glaucomas associated with other ocular and systemic disorders (many of which are discussed later in this chapter).
Although other nonglaucomatous eye conditions may share one or more findings with PCG, care must be taken to rule out other types of childhood glaucoma in each of these cases. For example, glaucoma may complicate uveitis and has been reported in the setting of MPS; corneal dystrophy; congenital anomalies, such as Peters anomaly; and megalocornea. Glaucoma has occurred coincidentally with congenital nasolacrimal duct obstruction (98).
Management
Medical Therapy
Definitive treatment of PCG is surgical in nature, with medical therapy playing an adjunctive role. Preoperatively, medications may help clear the cornea to facilitate angle surgery (especially goniotomy), and postoperatively, they may help control IOP until the adequacy of the surgical procedure has been verified. Medical therapy is also indicated in managing difficult cases in which surgery poses life-threatening risks or has incompletely controlled the glaucoma (7). In general, the same basic principles of medical therapy apply to the treatment of PCG as to adult glaucomas. One possible exception is the use of miotics, which paradoxically may increase the IOP by collapse of the trabecular meshwork because of the high insertion of uveal tissue into the posterior meshwork. (Dosages for children and special precautions are discussed in Chapter 40.) Many obstacles conspire against the success of chronic medical therapy for PCG, including inadequate IOP control, difficulties with long-term adherence, and the potential adverse systemic effects of protracted therapy.
Surgical Therapy
The primary surgical techniques are designed to eliminate the resistance to aqueous outflow created by the structural abnormalities in the anterior chamber angle. This “angle surgery” may be achieved with incisional surgery, by using an internal (goniotomy) or external (trabeculotomy) approach. Some surgeons prefer to perform a combined angle and filtration surgery (i.e., trabeculotomy and trabeculectomy) as the initial procedure; others use this technique after initial angle surgery has failed; and still others always perform filtration surgery only after angle surgery has failed (99–101). This discussion is limited to the concepts of management. (Details of the operative procedures are considered in Chapter 40.)
Goniotomy
Barkan (102) described a technique in which abnormal tissue (originally thought to be Barkan membrane) is incised under direct visualization with the aid of a goniolens. It is now believed that the incision is not through a membrane, but rather through the inner portion of the trabecular meshwork. This presumably relieves the compressive traction of the anterior uvea on the meshwork and eliminates any resistance imposed by incompletely developed inner meshwork.
Trabeculotomy
Harms and Dannheim (103) described a technique in which the Schlemm canal is identified by external dissection, and the trabecular meshwork is incised by passing a probe into the canal and then rotating it into the anterior chamber. One advantage of this procedure is that it can be performed in eyes with cloudy corneas, whereas goniotomy surgery requires visualization of the angle. Although some surgeons use the technique only in cases with corneal opacification, or when multiple goniotomies have failed, others prefer it as the initial procedure in PCG. In a modification of the earlier techniques, the Schlemm canal is cannulated for its entire circumference with a suture or an illuminated endoscopic probe, and then the encircling suture or endoscope is pulled, achieving a 360-degree trabeculotomy (104) (see also Chapter 40).
Goniotomy and trabeculotomy each have their advocates, and reported success rates vary considerably, with neither procedure having clear-cut superiority. Although goniotomy spares conjunctival tissues for possible later surgery, trabeculotomy can proceed even when corneal opacity precludes an angle view. (A more detailed comparison of the two operations is presented in Chapter 40.) With both procedures, success is related to the severity and duration of the glaucoma. The worst prognosis occurs for infants with elevated pressures and cloudy corneas at birth (primary newborn glaucoma). The most favorable outcomes are seen in infants who undergo surgery between the second and eighth month of life (primary infantile glaucoma), and the surgery then becomes less effective with increasing age (105). One study of long-term surgical outcome after trabeculotomy divided 71 children into groups of congenital glaucoma (i.e., existing before 2 months of age), infantile glaucoma (i.e., occurring between 2 months and 2 years), and juvenile glaucoma (i.e., after 2 years) and reported success rates with one or more trabeculotomies of 60.3% ± 5.9%, 96.3% ± 3.6%, and 76.4% ± 7.5%, respectively (106). Future studies may someday allow genetic identification of the patients with PCG who are more likely than others to benefit from angle surgery (107).
Other Glaucoma Procedures
When incisional angle surgery (e.g., goniotomies, trabeculotomies) has failed, alternatives include filtration surgery, glaucoma drainage-device surgery, and cyclodestruction, usually in that order. (Chapter 40 includes detailed review of published series using trabeculectomy with and without the use of antimetabolites, such as mitomycin C, to treat children with glaucoma.) Although older children with phakic eyes often achieve successful glaucoma control with this surgery (108), trabeculectomy is less likely to successfully control glaucoma in young infants because of their exuberant healing and scarring response. In addition, trabeculectomy in any child carries with it a lifetime risk for endophthalmitis (109–112).
Glaucoma drainage-device surgery also has a role in the management of infants and other children refractory to angle surgery and trabeculectomy. (Detailed discussion of implant surgery in children can be found in Chapter 40.) The Molteno, Baerveldt, and Ahmed implants have been used in children, with widely varying rates of success—from about 50% to greater than 90% (113–122). Glaucoma drainage-device surgery can successfully control glaucoma in children, although many patients need postoperative glaucoma medication therapy and repeated surgery. In one retrospective study, glaucoma drainage-device surgery had 5- and 10-year success rates of approximately 60% and 45%, respectively, in children with refractory PCG (123).
In contrast to trabeculectomy and glaucoma drainage-device surgeries, cyclodestructive procedures reduce the rate of aqueous production by injuring the ciliary processes; success is only modest (about 50%), results are often unpredictable, and complications occur frequently. Cyclodestruction nonetheless constitutes a valid means of attempting control of especially refractory cases of PCG after medical and other surgical means have been exhausted or have proved inadequate to the task. This modality may be reasonable to decrease aqueous production in eyes with elevated IOP despite patent glaucoma drainage-device surgery (Freedman SF, unpublished data). Cyclocryotherapy has been used to treat difficult childhood glaucomas for many years. Unfortunately, overall success (i.e., pressure control without severe visual loss or phthisis) has been poor (i.e., 30% success in a large series of children with advanced congenital glaucoma), and retreatment has been the rule (124). Transscleral cyclophotocoagulation with the contact Nd:YAG and diode lasers has reduced IOP in a fashion at least comparable to cyclocryotherapy in children with refractory glaucomas, and with a lower reported incidence of phthisis and hypotony (125–128). The endoscopic use of the diode laser for cycloablation has been applied to children (mostly with glaucoma in aphakia), with modestly encouraging results (129,130).
Penetrating Keratoplasty
Corneal cloudiness due to permanent scarring may persist after normalization of the IOP in some severe cases, prompting consideration of penetrating keratoplasty. Penetrating keratoplasty in young children is difficult, especially when the case is complicated by glaucoma and buphthalmos (131–134). These patients often do not fare well, with only 25% of eyes achieving 20/40 or better vision in one series (131). The most common postoperative complications are IOP elevation and graft failure. Although significant visual improvement can be achieved with penetrating keratoplasty (135), it is suggested that it be reserved for patients with severe visual disability whose glaucoma is well controlled (131). Optical iridectomy may be a less risky surgical compromise in eyes with central corneal opacity.
Postoperative Care, Prognosis, and Follow-up
The follow-up care of patients with PCG has several important facets. In the early postoperative period, close observation is required to maximize proper healing and odds of surgical success. In addition to IOP reduction, other clinical indicators of successful glaucoma control include clearing of corneal edema, reversal of optic nerve cupping, and even reduction in myopia in some cases (136). The IOP has also been related to postoperative visual capacity, with substantially better vision reported among those whose IOP remained no higher than 19 mm Hg in one series (105).
As with older patients, the target IOP for children with PCG should be guided by the severity of the optic nerve damage, with lower targets set for those eyes with lower central corneal thickness. In infants with healthy-appearing optic nerves (e.g., cup-to-disc ratio <0.5), a target pressure of about 20 mm Hg is often adequate; the stability of the optic nerve, corneal diameter, and refraction help confirm the adequacy of the IOP. Conversely, target pressures in the mid-teens are usually set for infants and children with severe pre-existing optic nerve damage. In selected cases, eyes can remain stable despite IOPs in the low- to mid-20s when optic nerve cupping and ocular expansion have been minimal. Medications should be used to further reduce the IOP after surgery if the target IOP is not met. As with adults, auxiliary techniques such as visual fields and serial optic nerve examination should be used and the target IOP altered if progression occurs despite IOP control at the previously set target level.
Even when the IOP is well controlled, a substantial number of children never achieve good vision. In previous studies, approximately one half of the patients had visual acuities of less than 20/50 (105,136,137). This reduction may result from persistent corneal changes or from irreversible optic nerve damage. A high percentage of patients, however, suffer amblyopia from the induced anisometropia, and it is critical that this condition be diagnosed early and managed appropriately.
Patients with PCG constitute a heterogeneous group, with overall IOP control achievable in more than 80% of cases. Although in rare cases primary infantile glaucoma has seemed to spontaneously remit (138,139), most untreated cases result in buphthalmos and blindness (3). The prognosis for control of PCG varies with the patient’s age at initial presentation and at surgery. Most favorable for IOP control with angle surgery are patients presenting after the first 2 months and within the first year of life (90%). Children presenting with glaucoma at birth or after the first year have a poorer prognosis for IOP control with angle surgery (50%) (7,140). Even children whose glaucoma is well controlled after surgical therapy (with or without adjunctive medical therapy) deserve lifelong follow-up. Loss of IOP control (reported in as many as approximately one third of cases (7)) may occur months or even decades after initial success with surgery and may be asymptomatic in the older child or young adult.
Congenital glaucoma is usually managed surgically, initially with a goniotomy or trabeculotomy. Visual loss due to amblyopia is common and should be aggressively treated. Lifelong follow-up is needed to guard against late loss of IOP control and to detect associated ocular problems such as corneal decompensation, cataract, and progressive optic nerve injury from inadequate IOP.
PRIMARY GLAUCOMAS WITH ASSOCIATED ABNORMALITIES (DEVELOPMENTAL GLAUCOMAS)
General Terminology
There have been considerable discussion and confusion regarding the lumping, splitting, and classifying of this large number of disorders. As the molecular genetics of anterior segment dysgenesis is further elucidated, the genetic classification of many of these disorders may supersede our current phenotypic descriptions. Nonetheless, the historical basis of our current classification system is worthy of further description here because management of many of these cases still rests firmly on their phenotypic presentation.
For simplicity, we consider all of the primary glaucomas that have associated ocular abnormalities to be forms of anterior segment dysgenesis. Based on the observation that most of the ocular and facial structures involved in these developmental disorders are of neural crest origin (67,141), the term neurocristopathies has been used as a unifying concept for diseases arising from neural crest maldevelopment (142,143). Hoskins and colleagues (144) advocated a shift away from eponyms and syndrome names for individual disorders and toward an emphasis on descriptive terminology. Noting that the trabecular meshwork, iris, and cornea are the three major structures involved in these conditions, they suggested using the terms trabeculodysgenesis, iridodysgenesis, and corneodysgenesis, or combinations thereof, to classify the developmental defects. In a recent review, Idrees and colleagues (145) consider the anterior segment dysgenesis to include not only Axenfeld–Rieger syndrome, iridogoniodysgenesis–iris hypoplasia, and Peters anomaly, but also PCG, sclerocornea, megalocornea, congenital hereditary endothelial dystrophy, and aniridia. Although most of these anterior segment dysgeneses are attributed to defects in neural crest migration or differentiation, aniridia is considered to be of non–neural crest origin. Many of the anterior segment dysgeneses also may occur with associated systemic abnormalities, further blurring the distinction between primary glaucomas with associated ocular abnormalities and those with associated systemic abnormalities (Table 13.1).
Although there is value in categorizing disorders on the basis of anatomic descriptions and mechanisms, the overlapping of manifestations and limited understanding of disease mechanisms make it difficult to apply such a system in all cases of developmental glaucomas with associated anomalies. There is often great phenotypic variability between individuals with the identical genetic cause for a disease; variation may even occur among members of the same family, making tissue classification inaccurate. For these reasons, traditional eponyms and syndrome names are retained with some suggested modifications for the purpose of discussion in this chapter. (For additional genetic information related to these conditions, see Table 8.1.)
Axenfeld–Rieger Syndrome
Terminology
In 1920, Axenfeld (146) described a patient with a white line in the posterior aspect of the cornea, near the limbus, and tissue strands extending from peripheral iris to this prominent line. Beginning in the mid-1930s, Rieger (147–149) reported cases with similar anterior segment anomalies, but with additional changes in the iris, including corectopia, atrophy, and hole formation. Some of these patients also had associated systemic developmental defects, especially of the teeth and facial bones (150,151). Axenfeld referred to his case as posterior embryotoxon of the cornea (146), and Rieger used the term mesodermal dysgenesis of the cornea and iris (149). Traditionally, these conditions have been designated by three eponyms: (a) Axenfeld anomaly (i.e., limited to peripheral anterior segment defects), (b) Rieger anomaly (i.e., peripheral abnormalities with additional changes in the iris), and (c) Rieger syndrome (i.e., ocular anomalies plus systemic developmental defects).
The similarity of anterior chamber angle abnormalities in the Axenfeld anomaly and the Rieger anomaly and syndrome led most investigators to agree that these three arbitrary categories represent a spectrum of developmental disorders (79,152,153). The overlap of ocular and systemic anomalies is such that the traditional classification is difficult to apply in all cases. Various collective terms have been applied to this spectrum of disorders, such as anterior chamber cleavage syndrome and mesodermal dysgenesis of the cornea and iris (149,152). As discussed earlier, the theories of normal development on which these names are based are no longer believed to be correct. The former collective term included the central ocular defects of Peters anomaly (discussed later in this chapter). Although some patients may have the combined defects of the Axenfeld anomaly or the Rieger anomaly and the Peters anomaly, the association is rare and the mechanisms of the two disorders differ in most cases.
The alternative term, Axenfeld–Rieger syndrome, was proposed for all clinical variations within this spectrum of developmental disorders and is now commonly used in the literature (79,154). This name retains reference to the original eponyms and does not depend on any theory of normal development, the understanding of which is still incomplete, nor does it require arbitrary subclassification of the clinical variations.
General Features
All patients with the Axenfeld–Rieger syndrome, irrespective of ocular manifestations, share the same general features: a bilateral, developmental disorder of the eyes; a frequent family history of the disorder, with an autosomal dominant mode of inheritance; no sex predilection; frequent systemic developmental defects; and a high incidence of associated glaucoma. The age at which the Axenfeld–Rieger syndrome is diagnosed ranges from birth to adulthood, with most cases recognized in infancy or childhood. The diagnosis may result from the discovery of an abnormal iris or other ocular anomaly, signs of congenital glaucoma, reduced vision in older patients, or systemic anomalies. In other cases, the condition is diagnosed during a routine examination, which might have been prompted by a family history of the disorder.
Ocular Features
Ocular defects in the Axenfeld–Rieger syndrome are typically bilateral. The structures most commonly involved are the peripheral cornea, anterior chamber angle, and iris.
Cornea
The characteristic abnormality of the cornea is a prominent, anteriorly displaced Schwalbe line. This appears on slitlamp examination as a white line on the posterior cornea near the limbus. The prominent line may be incomplete, usually limited to the temporal quadrant; in other patients, it may be seen for 360 degrees (Fig. 14.9). In some cases, the line can only be seen by gonioscopy, although rare cases with other ocular and systemic features of the syndrome may have grossly normal Schwalbe lines (155).
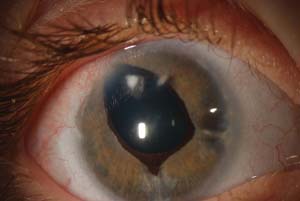
Figure 14.9 Right eye of a 6-year-old girl with Axenfeld–Rieger syndrome and congenital glaucoma. Note the prominent anteriorly displaced Schwalbe line, visible for almost 360 degrees. Ectropion uveae is also prominent. The pupil peaks toward the 11-o’clock position as a result of a glaucoma drainage device placed at 2 months of age. The tube also caused a focal cataract and an endothelial scar (before tube-shortening procedure). IOP is controlled and vision corrected to 20/40.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


